Sentieon发布RNAseq加速分析方案

RNAseq,即通过高通量测序技术进行转录组测序分析技术,作为研究RNA的表达水平以及表达差异基因的应用,在过去的十几年内迅速发展。而今,RNAseq在转录本变异检测,基因融合检测,可变剪切检测等场景均有大规模的应用。转录本变异检测,是指通过比较样本RNA序列和参考基因组对应序列,来寻找单碱基多态性和小片段的插入缺失,其结果大多用于治病位点的判断或性状相关的研究。融合基因是指两个或多个基因首尾相连,置于同一套调控序列控制之下,构成的嵌合基因,其表达产物为融合蛋白。在某些癌症中,融合基因的检测成为了重要的检测指标。
在数据分析方面,经过多年的探索与沉淀,业界针对不同的RNAseq应用逐渐产生了相应的主流分析方案。其中STAR作为一款经典的比对软件,在科研与临床的RNA测序数据分析中有着广泛的应用。相较于同样经典的Tophat2与HISAT2,STAR拥有更高的uniq mapping比例,且对lower-quality(包括more soft-clipped和错配碱基)比对有较高的容忍度,适用于更加复杂的分析需求,因此STAR成为Broad Institute RNA分析流程的最佳实践金标准。除此以外,还有包含了变异检测,定量分析,融合检测等其他分析模块共同被使用。
开源软件的一大问题就是速度较慢,耗时长。为克服这个问题,Sentieon开发了对应的加速模块,包括了比对步骤的Sentieon STAR、去重模块、处理RNA junction的模块和变异检测模块,以期缩短分析流程的耗时。
RNA变异检测
RNA变异(SNP和Indel)检测的重要性正在逐步被大家所认可。相比于DNA变异,RNA的变异对于异常蛋白的生成有着更加直接的意义,因此在临床上的应用也开始被大家所接受。相对的,加速分析的重要性也在凸显,因为这直接关系到受试者能否及时得到准确的检测结果。
与DNA变异检测类似,RNA的变异检测流程同样遵循业界的金标准GATK流程,包括了STAR比对,去重,RNA split的处理,Indel重比对(可选),BQSR,以及最终的变异检测等多个步骤。在本次的流程搭建中,我们利用Sentieon最新开发的STAR加速模块,与其他可用加速模块一起,完成了全流程的RNA变异检测流程的搭建工作。
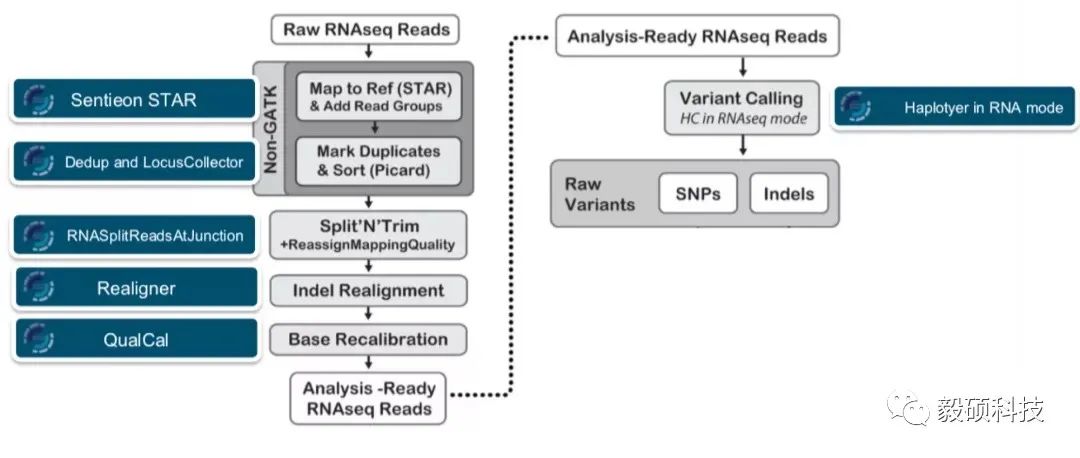
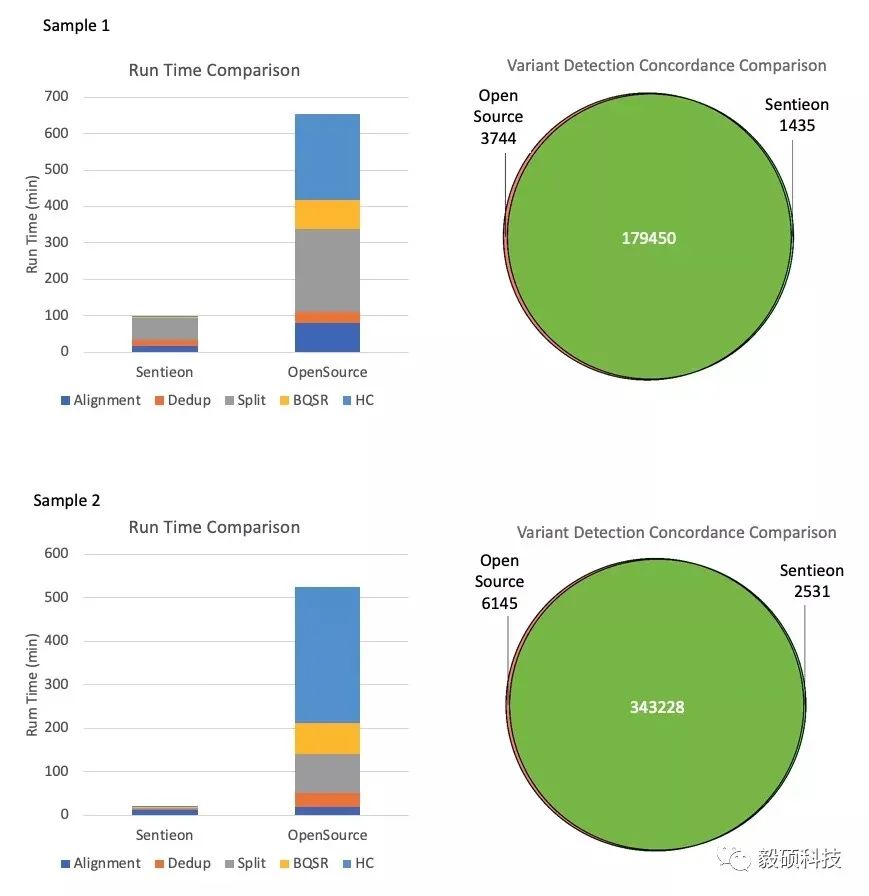
我们选取了2个RNAseq样本进行性能测试,运行包括原版STAR(2.7.8a版本)与GATK(4.2.0版本)在内的最佳实践流程,赋予同样的线程数再次运行搭建好的Sentieon流程,随后进行速度和一致性的比对。速度方面Sentieon各个模块的提速均比较明显,两个样本全流程的提速分别在6.6倍和23.9倍。两个流程的一致性在98.6-98.8%左右,主要区别来自于GATK版本号的不匹配。
RNA定量+基因融合
基因定量方面,我们使用SIRV样本作为测试样本。SIRV基因是Lexogen公司人工合成的7个基因,每个基因有多条转录本,共69个转录本,可用以检测可变剪切事件,并作定量内参使用。在这些转录本数据中我们选取了不同起始摩尔量的20个样本,分别使用原版STAR以及Sentieon STAR比对之后,使用Cufflinks2进行定量。我们共定量了155344个转录本,Sentieon与STAR流程的定量结果完全一致。由于这些样本的数据量较小(每个RNAseq样本8.9G左右,捕获样本数据1.3G左右),STAR在定量流程中所占比重也不太大,因此提速效果不是特别明显。
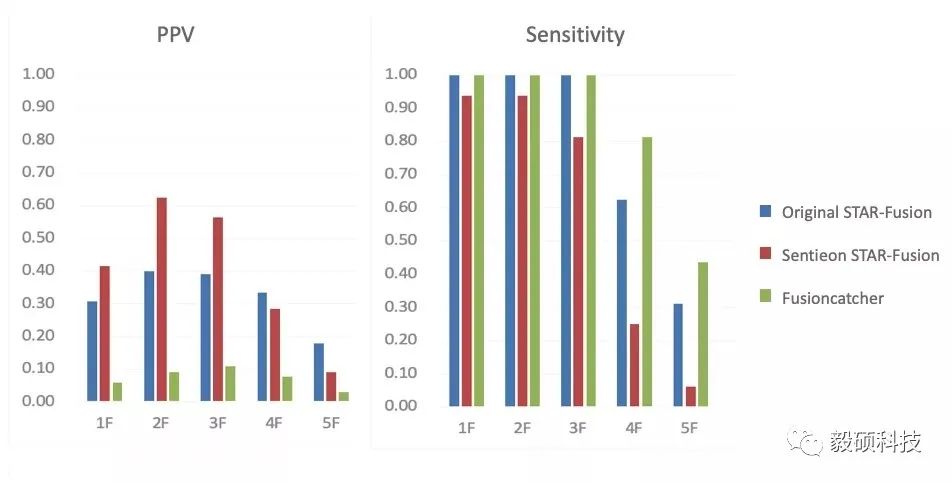
另外我们做了基因融合流程的搭建与检测,使用的参考标准品 (Seraseq FFPE NTRK fusion) 中包含了16个确定的基因融合事件,按照不同的比例与阴性样本混合之后生成5个样本(目标丰度0.23%-50%)作为评测样本。流程方面,我们测试了Sentieon STAR替换原版STAR进行测试,同时与Fusioncatcher进行比对。从结果来看,由于测试的STAR-Fusion中的STAR版本 (2.7.2b)与Sentieon STAR的匹配版本(2.7.8a)不同,两个流程的检出率与特异性也略有差异,总的来说Sentieon流程在PPV上有较好的表现,但在Sensitivity上略低。
方案总结
在本次方案合作中,Sentieon提供模块组件,福君团队搭建并测试了RNA变异检测流程,纳昂达团队负责了RNA定量与基因融合的相关部分。经过真实数据的评测,我们通过数据展示了Sentieon流程在RNAseq的三项不同应用之中的性能提升,希望能够为业界选择合适的RNAseq分析流程提供参考。

- 点赞
- 收藏
- 关注作者
评论(0)