QUARK的增强版C-QUARK问世,有效提升蛋白质结构从头预测精度


2021年8月18日,密西根大学张阳教授团队在Nature Communications上发表论文“Improving fragment-based ab initio protein structure assembly using low-accuracy contact-map predictions”。基于序列的接触预测在辅助非同源蛋白质结构建模方面具有相当大的前景,但这种方法通常需要许多同源序列以及足够数量的正确接触才能实现蛋白质的正确折叠。作者研究开发了一种方法:C-QUARK,它集成了多个深度学习方法和基于共进化分析预测得到的接触图,实现基于副本交换蒙特卡罗方法片段组装过程,是QUARK的增强版。该方法在第13次CASP赛事(蛋白质结构预测领域的奥利匹克竞赛)中FM(无模板)蛋白质结构建模盲测中相比其他基于接触图的非同源蛋白质建模方法更具有显著优势,证明了C-QUARK即使在同源序列少或接触预测精度不高时也可以实现蛋白质三维结构的有效预测。
1.研究背景
蛋白质结构从头预测通常是指在PDB(蛋白质数据库)中不使用同源模板即可对蛋白质结构进行建模的方法,在过去几十年中一直吸引着人们的研究兴趣。十年前,包括Rosetta和QUARK在内的几个先进pipelines在CASP(critical assessment of protein structure prediction)实验中开创性地为长度超过100个残基的挑战性目标生成了正确的折叠,但这些从头折叠方法仍远不尽如人意。克服从头算建模局限性的有效方法之一是在折叠模拟中结合长程接触,但仍需要足够高精度的接触预测,以便建模精度不会因为太多错误预测的接触而受到影响。直接耦合分析(DCA)和深度神经网络学习技术的引入实现了接触预测精度的飞跃。但尽管在接触预测方面取得了显著进展,除非预测接触与折叠模拟有效集成,否则当前的从头建模协议无法充分发挥其潜力。特别是,当同源序列的数量较少以及基于序列的接触预测的准确性较低时,如何平衡有噪声的接触图与先进的折叠模拟力场以构建正确的从头结构折叠仍然是一个重要且具有挑战性的问题。
本文研究开发了一个接触引导从头折叠程序,C-QUARK。为了系统地探索接触图预测的能力,特别是那些精度较低的接触图预测,以改进从头折叠,该研究将接触约束与基于QUARK的折叠模拟结合了起来。该pipeline 在CASP目标和单独的大规模测试数据集上进行了严格的基准测试,结果显示,与QUARK以及其他先进的结构建模方法相比,C-QUARK在建模长距离和非同源目标方面显示出显著优势。
2.介绍
C-QUARK是基于QUARK这一顶级从头蛋白质折叠模拟程序之一建立的,其算法主要分为五个步骤:通过DeepMSA生成多序列比对、基于深度学习的接触图预测、片段构建、接触图引导的副本交换蒙特卡罗折叠模拟、模型选择与细化。C-QUARK从全基因组和宏基因组序列数据库的多序列比对(MSA)收集开始,其中两种类型的接触图是通过基于深度学习和协同进化的预测器创建的。接下来,从不相关的PDB结构中收集具有连续序列长度(1-20 AA)的结构片段,并在由基于知识的能量项、基于其距离分布从结构片段中收集的残基间接触以及基于序列的接触图预测组成的复合力场的指导下,通过副本交换蒙特卡罗(REMC)模拟将其用于组装全长结构模型(图1)。然后,通过SPICKER对模拟轨迹中的构象进行聚类,识别对应最低自由能状态的簇,并通过分子动力学结构细化方法FG-MD进一步优化簇心,获得最终的结构模型。
与QUARK相比,C-QUARK有三个主要实现项,包括:(1) 多序列比对生成工具DeepMSA,用于构建谱和预测接触图;(2) 基于深度学习和协同进化的接触预测模块,用于残基接触图预测、组合和选择;(3) 开发并精心训练了接触势能项来平衡接触势能与其他能量项(包括固有知识和物理势能),以指导结构组装模拟。
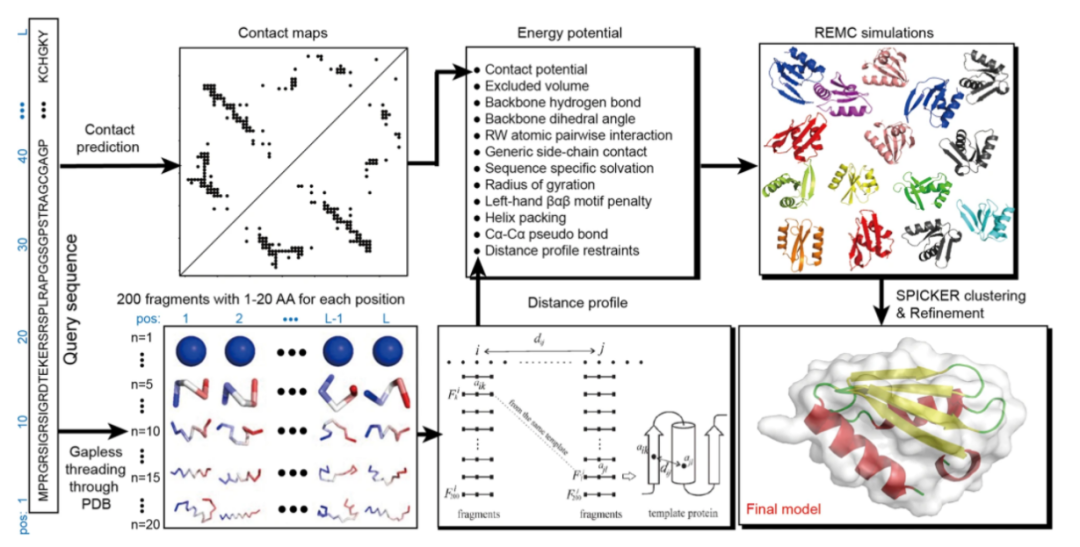
图1.C-QUARK流程图
3.结果
C-QUARK在从头结构预测中显著优于QUARK
由于C-QUARK和QUARK之间的主要区别在于前者程序中包含了接触约束,因此对这两个程序进行基准测试可以检验接触图在蛋白质从头折叠中的有效性。作者从PDB收集了一组包含247个非冗余单域蛋白质的集合,其分辨率优于3Å,长度在50–300个残基之间。表1总结了测试结果,其中C-QUARK的第一个模型的平均TM-score(0.606)比QUARK (0.423)高出43%。表1还列出了前五名模型中最佳模型的结果,其中C-QUARK再次显著优于QUARK,其平均TM-score(0.629)比后者(0.468)高34%。这里,TM-score是用于评估模型预测结构和原生结构之间的结构相似性(即模型预测准确性)的指标,取值介于0到1之间,TM-score> 0.5则表示模型预测的结构与真实结构相似。
此外,数据显示,在247个测试蛋白上,C-QUARK有224个测试蛋白生成的模型比QUARK生成的模型具有更高的TM-score。如果用正确的结构折叠来计算, C-QUARK的第一个模型有186个(75%)蛋白质被正确折叠,而QUARK只有71个(29%)蛋白质被正确折叠。C-QUARK正确折叠了46% QUARK无法折叠的蛋白质,表明了接触约束在C-QUARK蛋白质结构从头折叠的过程中起了主要的积极影响。
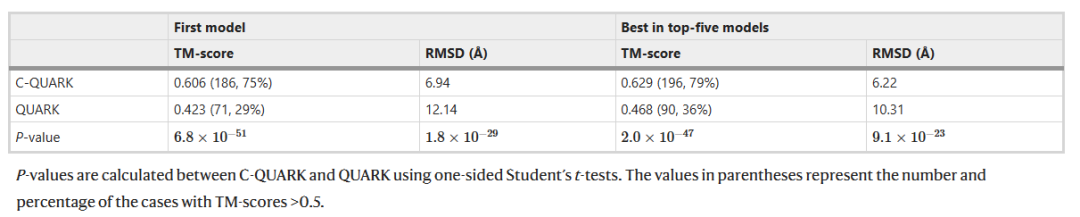
表1.C-QUARK与QUARK对247个测试蛋白质的预测性能
为了评估C-QUARK对不同蛋白质类型进行建模的能力,作者根据测试目标的二级结构组成将测试目标分为三类,即α、β和α-β蛋白(图2)。对于建模相对容易的α-蛋白,C-QUARK的正确折叠率(64种α蛋白的52种)几乎是QUARK(64种α蛋白的24种)的两倍,而对于β和α-β类型的蛋白质,C-QUARK的折叠成功率比QUARK高约三倍。其中β-蛋白建模精度的提高尤其令人振奋,因为β-蛋白通常具有以长程接触图为特征的复杂拓扑结构而导致其从头建模一直相当困难,其从头折叠程序的固有力场通常难以捕捉由微妙的氢键网络形成的这种长程相互作用。C-QUARK结合长程残基间接触预测有效地捕捉到了这种相互作用,并显著改善了具有复杂β折叠拓扑的目标的折叠性能。
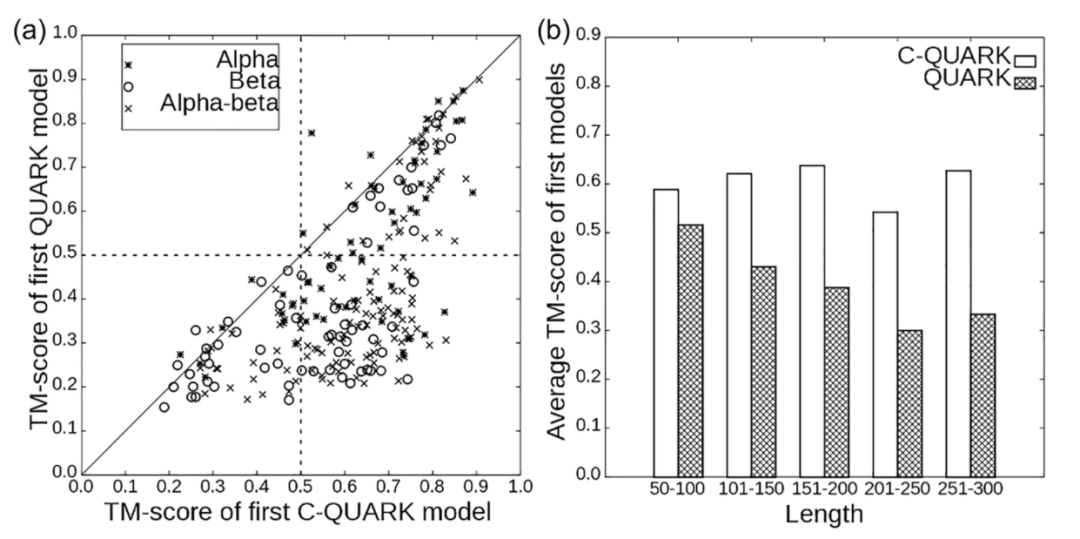
图2.C-QUARK和QUARK在247个测试蛋白上的比较结果。a.C-QUARK与QUARK对不同蛋白质类别的第一个模型的TM-score。b .在不同的蛋白质长度区间,C-QUARK和QUARK的平均TM-score。
对于缺乏同源序列和高精度接触的目标,C-QUARK 明显优于其他接触引导折叠方法
作者将C-QUARK的性能与其他三个主要基于预测的接触或距离构建结构模型的程序进行了比较,包括CNS、DConStruct(v1.0)和trRosetta(v1.0)。值得注意的是,在有效序列数低(Nf < 15)或接触图精度低(< 30%)的59个目标中,C-QUARK为24个目标(41%)生成了正确的折叠,而CNS(DConStruct)仅为4(4)个获得了正确的折叠。由于低Nf MSAs的接触预测一直是接触引导从头建模的瓶颈,因此C-QUARK在为这些具有挑战性的目标生成正确模型方面,其成功率的显著提高尤其令人鼓舞。同时,C-QUARK在这59个目标的TM-score (0.428)也明显高于QUARK (0.348),表明尽管精度相对较低,接触图预测仍然有助于折叠。
QUARK优于其他基于接触的折叠程序,这主要是因为其在结构组装模拟中综合力场的帮助。C-QUARK的成功应归功于预测的残基-残基接触与固有力场和结构组装模拟过程之间的相互作用。
C-QUARK 在CASP13目标上的性能测试
为了直接将 C-QUARK 与其他最先进的结构预测程序进行比较,C-QUARK 作为“QUARK”服务器参与了第13次结构预测关键评估(CASP13)实验。作者分析了C-QUARK在64个CASP13 FM(free modeling)、FM/TBM(free modeling/template-based modeling)和TBM-hard(template-based modeling-hard)目标上的性能。这些目标具有挑战性,因为同源模板不存在或难以从PDB库中检测到。根据64个CASP13目标的实验结构,C-QUARK的平均GDT_TS(CASP评估员使用的标准分数)高于所有其他p值<0.05的参与服务器的平均GDT_TS。尤其是在TBM-hard和FM类别中,C-QUARK分别比第二好的方法好4%和5%。而对于FM/TBM目标,BAKER-ROSETTASERVER (60.58) 略好于C-QUARK (58.94),但差异并不显著。
4.总结
在这项研究中,作者开发了基于(稀疏)接触图引导的蛋白质结构从头预测算法C-QUARK,它显示出对在PDB没有同源模板的“hard”蛋白质建模的能力显著提高。虽然C-QUARK pipeline建立在该领域顶级从头建模程序之一的QUARK平台上,但当基于序列的接触预测被纳入时,平均TM-score提高了43%。重要的是,C-QUARK生成正确折叠的总体成功率约为75%,是QUARK (29%)的2.6倍,表明接触图预测在改进从头结构建模中的重要性。此外,C-QUARK还显示出一致的折叠长度大于150个残基的中型到大型蛋白质的能力,这一直是几十年来从头建模领域的限制之一。
参考文献
Mortuza, S.M., Zheng, W., Zhang, C. et al. Improving fragment-based ab initio protein structure assembly using low-accuracy contact-map predictions. Nat Commun 12, 5011 (2021).
https://doi.org/10.1038/s41467-021-25316-w
代码\数据链接:
https://zhanggroup.org/C-QUARK/
https://github.com/jlspzw/C-QUARK
文章来源: drugai.blog.csdn.net,作者:DrugAI,版权归原作者所有,如需转载,请联系作者。
原文链接:drugai.blog.csdn.net/article/details/120574364

- 点赞
- 收藏
- 关注作者
评论(0)