Chem. Sci. | 3D深度生成模型进行基于结构的从头药物设计


本文介绍来自北京大学来鲁华教授课题组发表在Chemical Science上的文章“Structure-based de novo drug design using 3D deep generative models”。在这项工作中,作者介绍了一种基于深度学习的新型药物设计方法DeepLigBuilder,可以在靶标蛋白结合位点生成三维(3D)分子结构。模型中配体神经网络(Ligand Neural Network,L-Net)生成在化学上和构象上有效的,具有高度药物相似性的3D分子。然后,L-Net和蒙特卡洛树搜索(MCTS)结合,完成基于结构的药物从头设计任务。DeepLigBuilder将深度生成模型与原子级相互作用评估相结合,为基于结构的新药设计和先导物优化提供了最先进的模型。
1
介绍
新药物发现的核心任务便是寻找具有良好生物活性和成药性的分子,但它受制于规模巨大且复杂的化学空间。最近,各种深度生成模型被引入到从头设计领域。传统方法为保证输出分子的实用性,通常需要专家制定规则,而深度生成模型大多是“无规则”的,可以完全以数据驱动的方式训练,对专家知识的需求最小。基于SMILES和基于图的语言模型、VAE,GAN是最常使用的深度学习架构。
在药物发现过程中,已经开发了各种方法来满足不同的需求,如基于性质、基于靶点、基于药效团和基于骨架的分子设计方法。然而,与传统方法直接在结合口袋的3D结构内部构建3D配体不同,大多数基于深度学习的方法只生成一维(1D)(smile)或二维(2D)(Graph)分子表示,没有使用目标的结构和相互作用信息。因此,这些方法大多依赖于基于配体的目标,这可能会受到与训练集中的配体相关的偏差的影响。这也使得模型难以应用于生物活性数据有限的病例。而基于结构的信息为优化配体与靶点之间的相互作用提供了直接指导,降低了对现有生物活性信息的依赖。因此,将3D条件纳入深层分子生成模型是非常可取的。
2
方法
作者实现了以一种新的从头分子设计模型DeepLigBuilder,它同时享有了传统方法和深度学习方法的优势,使用深度生成模型直接在3D结合口袋内构建和优化配体的3D结构。作者将这个目标分为以下两个任务。首先训练一个可以生成具有有效3D结构的类药物分子;其次,将基于靶标的信息引入模型中,从而得到具有预测良好的结合亲和力的分子。
L-Net介绍
针对第一个任务,作者采用了一种专门用于生成3D类药物分子的图形生成模型L-Net。L-Net有两个对其性能很重要的特征。首先,它由一个结合了图池化(graph pooling)和旋转协方差等特征的新的图卷积结构构建的,增加了网络的感受野的大小,同时提高了训练和采样的效率。其次,L-Net使用了一种新的方案进行训练,使其能抵抗生成过程中的3D误差。因此,该模型可以生成化学性质正确、构象有效和高度药物样的分子。与G-SchNet相比,L-Net实现了显著更好的化学有效性,同时保持了生成构象的质量提高。
L-Net使用状态编码器和策略网络,通过迭代精炼现有结构生成3D分子。如图1a,在每个步骤中,状态编码器首先用来分析现有的结构,并将信息编码成连续的表示,然后由策略网络使用该表示来决定分子应该如何编辑。策略网络决定应该添加多少原子到分子中,每个新原子和键的类型,以及新原子的3D位置。
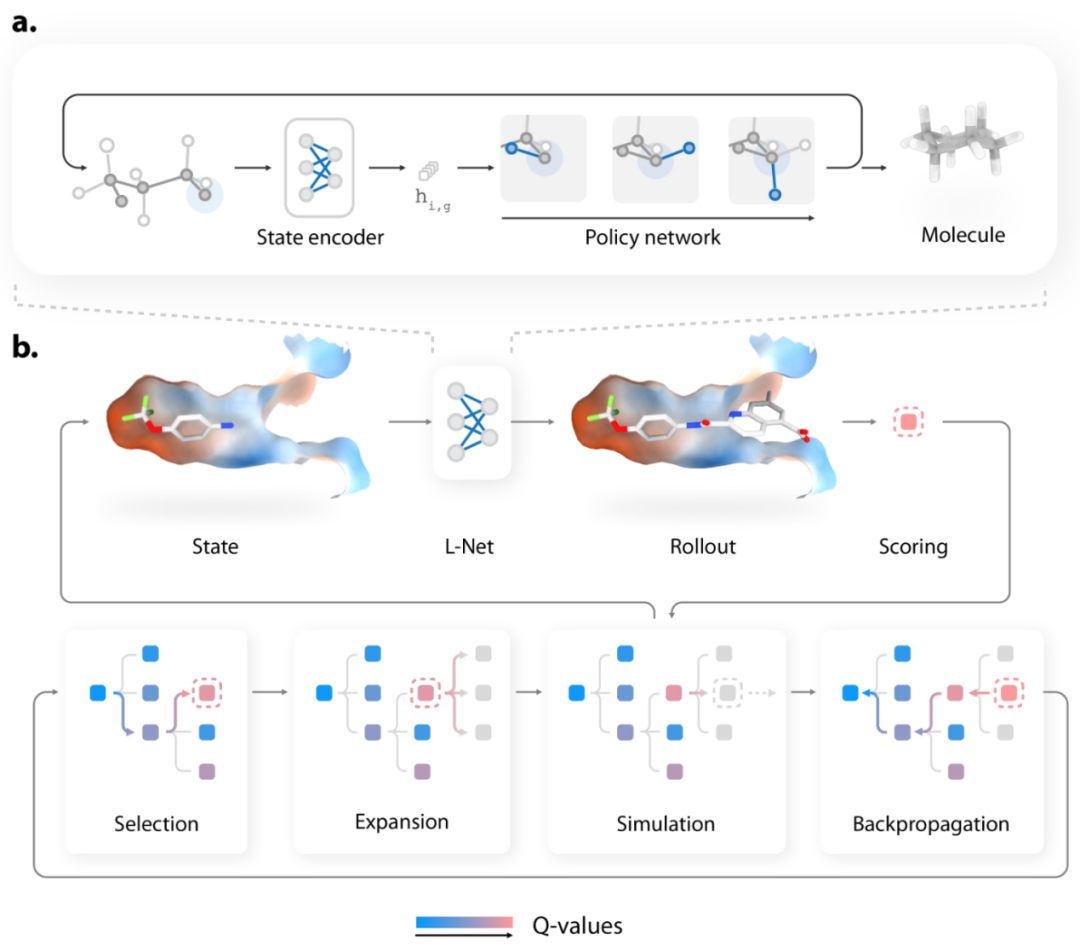
图1. DeepLigBuilder架构
作者设计了一个类药物数据集(QED>0.5),包含从ChEMBL中过滤得到的100万个由RDKit生成构象的分子。L-Net使用了一整套常见的原子类型({C, H, O, N, P, S, F, Cl, Br, I}),覆盖了ChEMBL数据集中的大多数类药物分子。训练期间,模型需要“模仿”数据集中的分子,具体来说,就是为每个分子创建一个“专家轨迹”来形成其结构。
利用L-Net进行基于结构的分子设计
为完成第二项任务,作者将L-Net与MCTS相结合,在蛋白结合口袋内直接生成具有高预测亲和力的配体,称该方法名为DeepLigBuilder。MCTS是一种广泛应用于强化学习的技术,可以直接在结合口袋内对分子进行优化。作者调查表明,这是第一次将3D生成模型与MCTS结合来解决基于结构的药物发现(SBDD)相关问题。
如图1b,展示了L-Net结合MCTS优化蛋白结合口袋内药物样分子的流程。MCTS通过迭代构建搜索树来找到奖励函数的有希望的解。树中的每个节点代表分子生成过程中的一个中间状态。在每次迭代中,模型首先从搜索树中选择一个有希望的状态(selection),枚举该状态的可能操作(expansion),并执行rollout以生成其余的分子结构(simulation)。为该结构收集奖励,信息通过树反向传播,更新每个节点的q值。其中,作者使用了smina 提供的对接得分作为奖励函数。
MCTS负责寻找高结合亲和力的分子,而L-Net用于促进结构变得有效、类似药物、易于合成和多样化。
3
结果
Net分子生成
作者设定了如下的一组评估指标来综合评估L-Net的性能,特别强调了3D结构质量:
-
生成在化学上有效的化合物的能力;
-
生成样本的多样性;
-
正确学习重要分子性质分布的能力;
-
L-Net是否能正确为训练集的化学空间(2D和3D)建模;
-
生成高质量构象的能力。
图2显示了L-Net的定量评估结果,每个指标都是使用L-Net中随机生成的50,000个样本进行计算的。高达94.3%输出有效性表明模型能够正确地学习拓扑结构的化学约束。通过计算 MMFF94s 力场优化前后结构之间的 RMSD 值来评估生成的分子构象的质量。结果表明,L-Net在性能上比G- SchNet(3D生成任务中之前最先进的模型)略胜一筹。
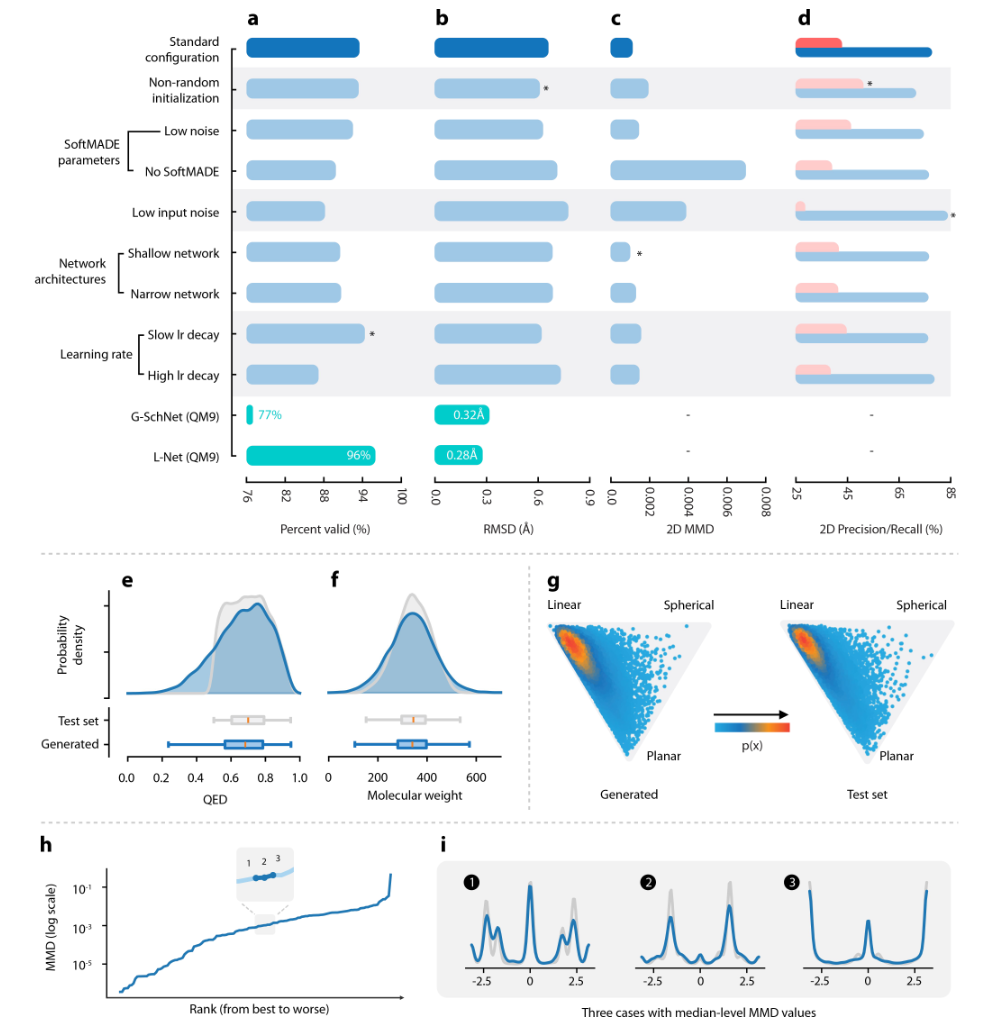
图2.L-Net的定量评估。a-d: 在四种方式(有效输出百分比、优化后的RMSD值、2D MMD、2D preceision (pink) and recall (blue))上测量的L-Net的性能;e-f:QED和分子量分布;g:使用NPR描述符可视化的分子形状分布;
使用DeepLigBuilder进行基于结构的分子设计
作者使用DeepLigBuilder设计了病毒SARS-CoV-2的主要蛋白酶(Mpro,也称为3CL蛋白酶)的潜在抑制剂。首先研究了DeepLigBuilder基于现有拟肽共价抑制剂的片段进行先导物优化的能力。然后在非共价抑制剂的新设计上测试了它的性能,目的是生成具有新型支架的高效抑制剂。
作者使用DeepLigBuilder 模拟MI-23设计过程,检测模型是否能生成具有良好药物相似性和高预测结合亲和力的分子。利用Mpro与MI-23配合物的结构进行分子生成,只保留P1片段(图3a,蓝色)作为生成的起始点。实验结果(图3.b-h)发现,给定一个已知的特权片段,DeepLigBuilder能够生成具有良好的预测结合亲和力、合理的药物相似性和与已知抑制剂相似的结合特征的分子。这些属性证明了DeepLigBuilder在引导优化问题方面的实际适用性。
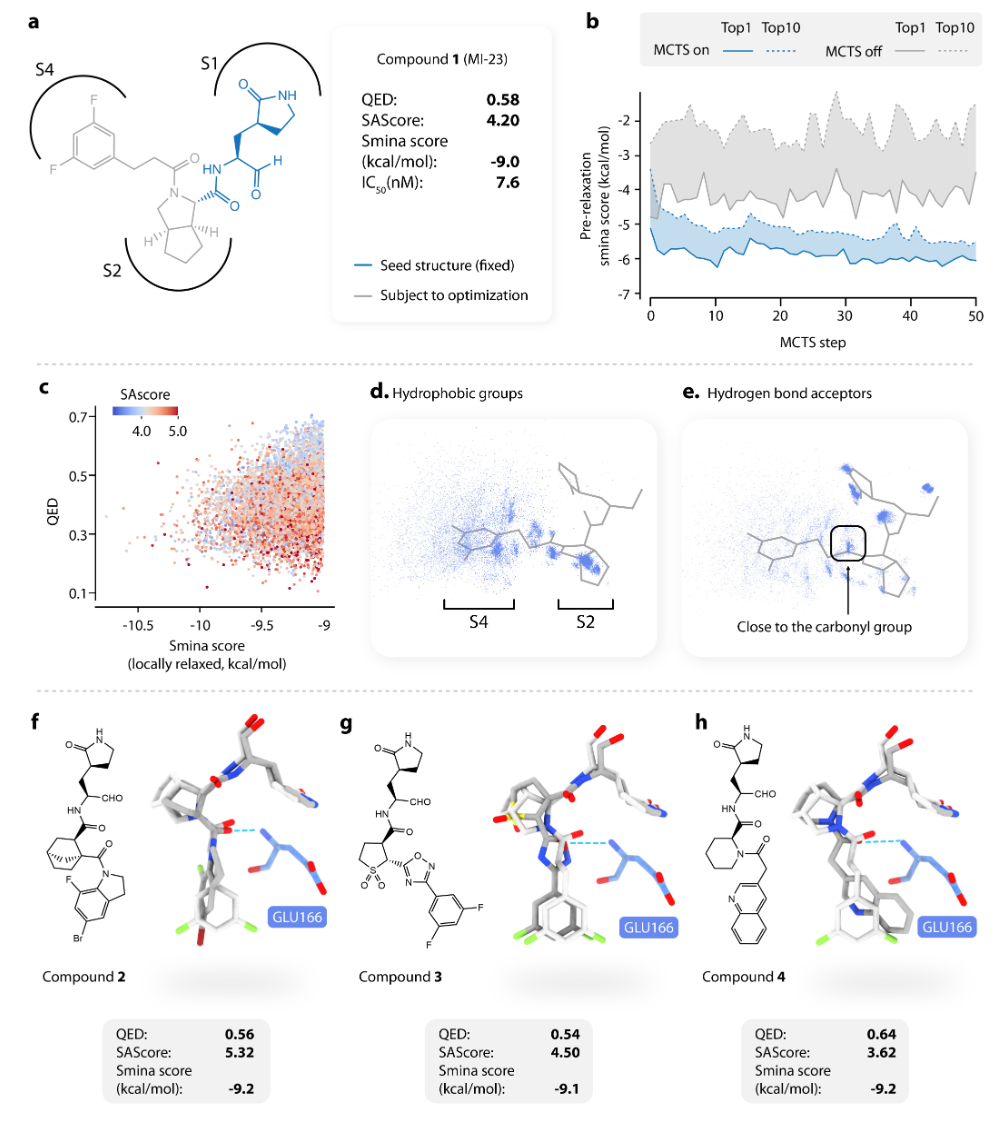
图3.使用DeepLigBuilder进行先导物优化
接着,作者使用DeepLigBuilder设计针对Mpro的非共价抑制剂。图 4b 显示了一种具有高生物活性的分子(化合物 5,IC50=0.128μM)。作者只使用化合物5中含有三个原子的小片段作为生成种子(图4b,蓝色),目标是生成具有新型骨架的分子。实验结果(图4d-f)表明在生成的分子中可以观察到蛋白-配体相互作用的重要药效团特征。
除此之外,作者还分析了生成分子的化学骨架,选择3个与化合物5相比QED、SAscore和smina评分提高的化合物进行进一步分析。实验结果展示了DeepLigBuilder发现与靶蛋白新相互作用的能力。但是这些设计的化合物是否能有效抑制Mpro还需要进一步的实验研究。
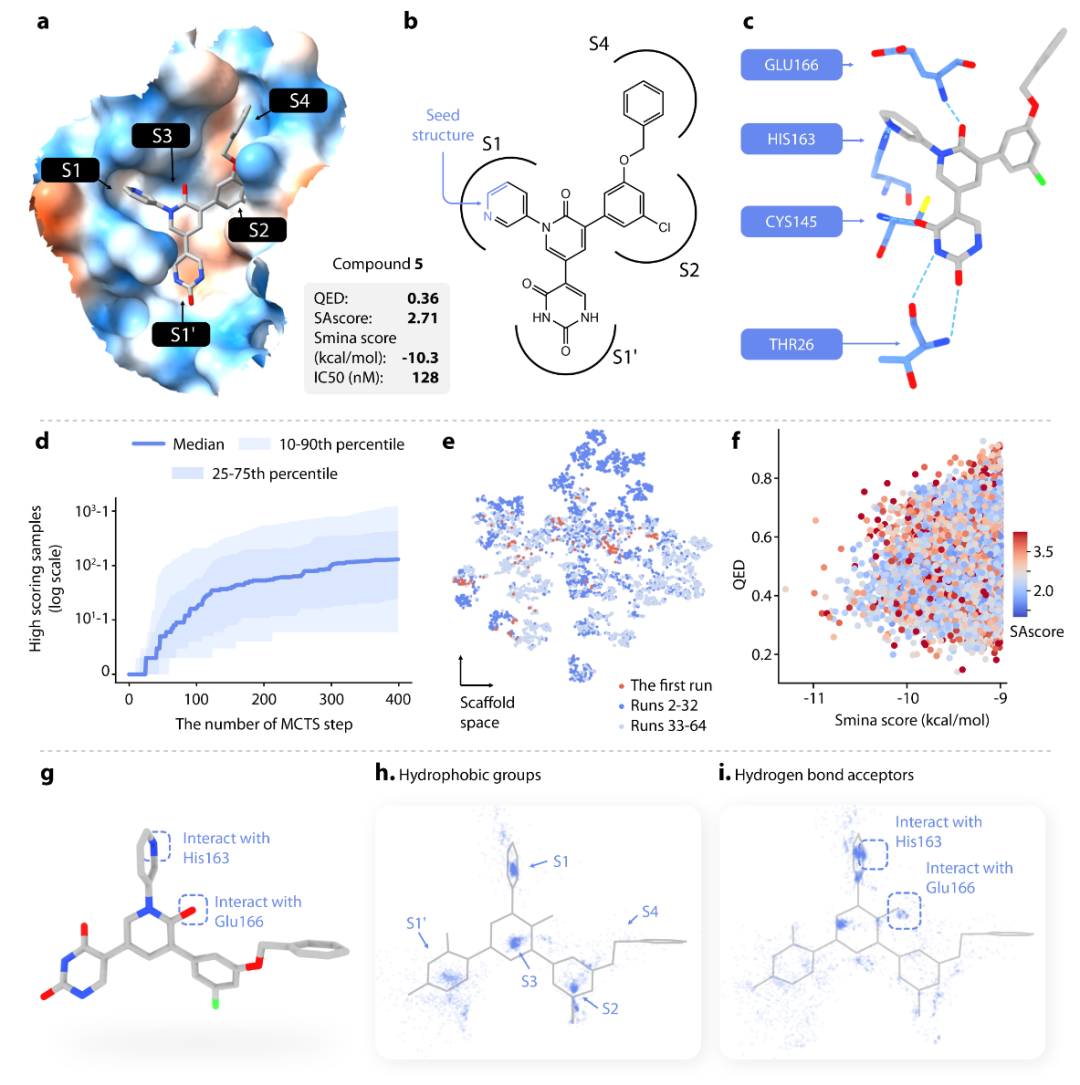
图4.使用DeepLigBuilder进行分子生成
4
总结
作者开发了一种基于深度学习的新型药物设计方法DeepLigBuilder,可在靶蛋白结合位点生成3D分子结构。模型使用L-Net生成有效的3D类药物分子,使用MCTS搜索强结合分子,可以直接输出类药物分子的3D和拓扑结构,而不需要额外的原子位置或键序推断。DeepLigBuilder能够生产具有新型化学结构、高预测结合亲和力的有前途的类药物化合物,捕获已知抑制剂的重要药效团特征,用于先导物优化和从头生成任务。
DeepLigBuilder的独特之处在于,它直接操作3D分子结构,并在同一时间直接使用MCTS在结合口袋内优化分子的拓扑和3D结构。由于它能够直接操作3D结构,DeepLigBuilder更加灵活,可以轻松实现更高级的功能,如在空间位置锚定生成分子或基于特权3D片段执行生成。
参考文献
Li, Y.; Pei, J.; Lai, L. Structure-Based de Novo Drug Design Using 3D Deep Generative Models. Chem. Sci. 2021.
https://doi.org/10.1039/D1SC04444C.
文章来源: drugai.blog.csdn.net,作者:DrugAI,版权归原作者所有,如需转载,请联系作者。
原文链接:drugai.blog.csdn.net/article/details/120559053

- 点赞
- 收藏
- 关注作者
评论(0)