Nat. Commun. | 可多层次预测多肽-蛋白质相互作用的深度学习框架


本次报道的论文来自清华大学的曾坚阳老师团队发表在nature communications上的A deep-learning framework for multi-level peptide–protein interaction prediction。文章提出了一个可多层次预测多肽-蛋白相互作用的深度学习框架(CAMP)。该模型包括二元多肽-蛋白相互作用预测和相应的多肽结合残基鉴定,通过综合测试指标表明,CAMP可以成功地捕获多肽和蛋白质之间的二元相互作用,并识别参与相互作用的多肽上的结合残基。此外,CAMP在二元多肽-蛋白相互作用预测方面优于其他最先进的方法。模型可以作为多肽-蛋白相互作用预测和鉴定多肽中重要结合残基的有用工具,从而促进多肽药物的发现过程。
1
研究背景
多肽通过与多种蛋白质相互作用,参与许多细胞过程,如程序性细胞死亡、基因表达调控和信号转导,在人类生理中发挥重要作用。目前有两种主流的蛋白质-配体相互作用的预测方法,分别是基于序列的和基于结构的方法。然而,现有的方法主要集中于识别蛋白质表面的多肽结合残基,基于序列的方法无法进行大规模计算,基于结构的方法需要用到三维结构信息,但计算获得三维结构信息非常昂贵和耗时。在这里作者提出了CAMP,一种深度学习框架,用于同时预测多肽-蛋白相互作用(pepPIs)和识别多肽序列的结合残基。
2
模型介绍
CAMP利用两个多通道特征提取器来分别处理输入的多肽-蛋白对的特征轮廓。每个提取器包含一个数值通道和三个分类通道。数值通道用于提取预先定义的密集特征(即蛋白质位置特异性评分矩阵(PSSM)和蛋白质和多肽序列中每个残基的内在无序趋势)。每个分类通道包含一个self-learning word embedding层,它具有输入多肽或蛋白质的分类特征之一(即原始氨基酸、二级结构、极性和亲水性特性)。接下来,CAMP利用两个卷积神经网络(CNN)模块,分别提取多肽和蛋白质的隐藏特征。此外,CAMP采用self-attention机制来学习残基之间的长期依赖关系,以及蛋白质和多肽的单个残基对最终相互作用预测的贡献。最后,CAMP结合了所有提取的特征,并使用三个完全连接的层来预测给定的多肽-蛋白对之间是否存在相互作用。
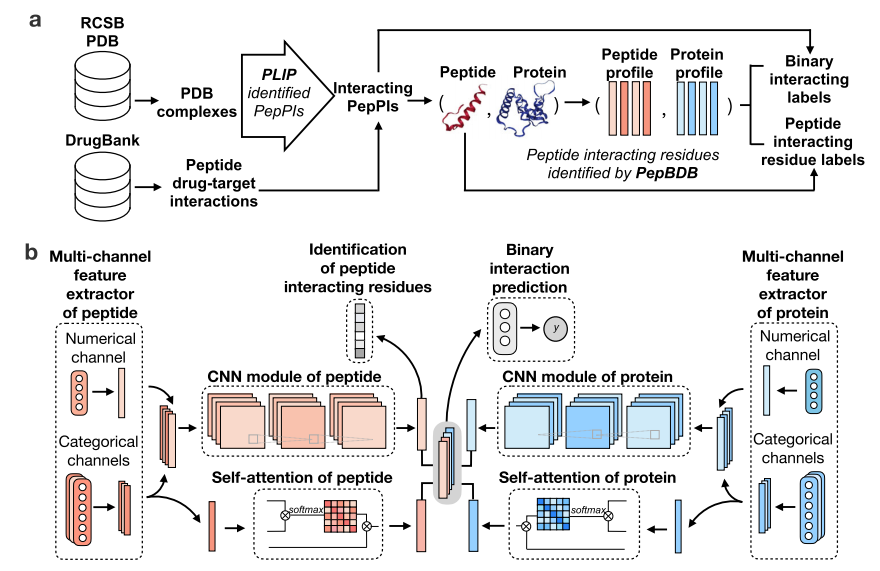
图1. CAMP流程图。a. 数据收集和标签提取的工作流程 b . CAMP的网络架构
3
结果
CAMP在二进制交互预测方面优于baseline方法
pepPIs的二元分类是CAMP的主要目标。作者比较了CAMP的分类性能与其他最先进的baseline方法进行比较,所有的预测方法均通过交叉验证在基准数据集上进行评估。图2显示,CAMP始终优于最先进的baseline方法,在ROC特征曲线下面积(AUC)和精度召回率曲线下面积(AUPR)方面分别增加了10%和15%。此外,作者还注意到,在“New Peptide Setting”的数据划分下的模型性能似乎比在其他设置中更好,这可以解释为在基准集中的多肽之间的相似性少于蛋白质,因此基于相似性聚类后,训练和测试集中的多肽分布没有太大变化。这些测试结果表明,在所有交叉验证设置下,CAMP都可以比baseline方法获得更好的性能和鲁棒性。
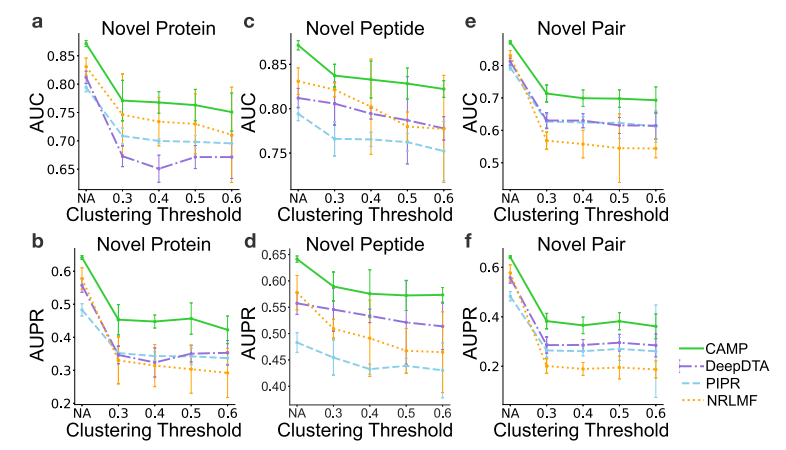
图2. 通过三种设置下的交叉验证,CAMP和baseline模型的AUC和AUPR
描述多肽上的结合残基的新见解
作者设计了一个监督预测模块来从多肽序列中识别结合残基,首先利用来自PepBDB的相互作用信息构建了一套合格的多肽结合残基标签。在这些监督信息的支持下,CAMP的平均AUC为0.806,Matthes相关系数(MCC)为 0.514(图3a,3b)。作者为了进一步证明CAMP在结合残基预测中的性能,还选择了4个代表性病例,并将预测残基与真实的相互作用残基进行比较(图3c,3d,3e,3f)。测试结果表明,CAMP可以准确地预测结合残基,从而为进一步理解多肽与其伴侣蛋白的相互作用机制提供可靠的证据。
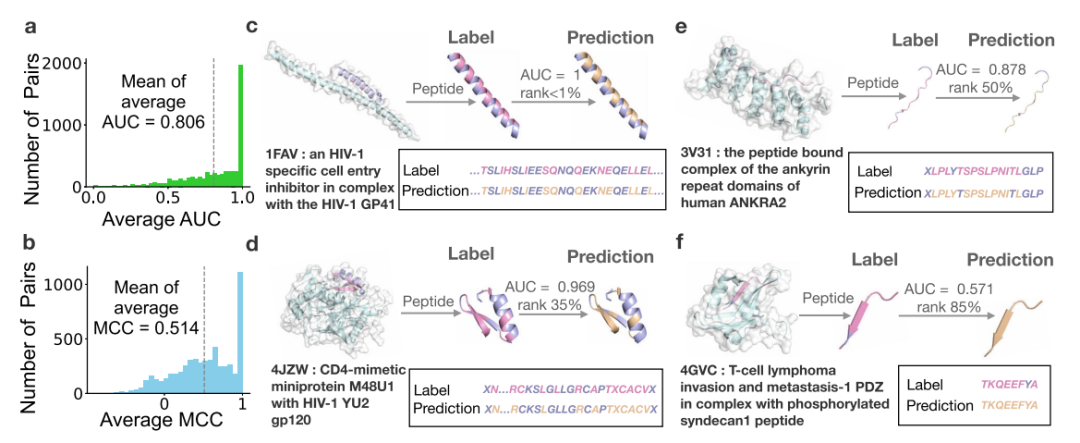
图3. 通过5倍交叉验证,对CAMP对基准数据集上多肽结合残基鉴定的性能评价
CAMP在其他基准数据集上的通用性
首先在来自PDB的额外独立数据集上评估了CAMP,并遵循与构建之前的基准数据集相同的策略。为了证明CAMP在二进制交互预测方面的鲁棒性,作者评估了CAMP和基线模型在不同正负比的测试数据集的多个不同的变化上的性能。每个模型首先在完整的基准数据集上进行训练,然后使用一个集成版本(即来自五个模型的平均预测)对额外的测试数据集进行预测。图4a和4b显示,CAMP在所有场景下都取得了最好的结果,表明CAMP的性能优于baseline方法。同时,还评估了CAMP对多肽结合残基鉴定的预测结果。从PepBDB中获得了多肽序列的注释结合残基。图4c和4d显示,CAMP能够在上述额外的数据集上保持其预测能力。
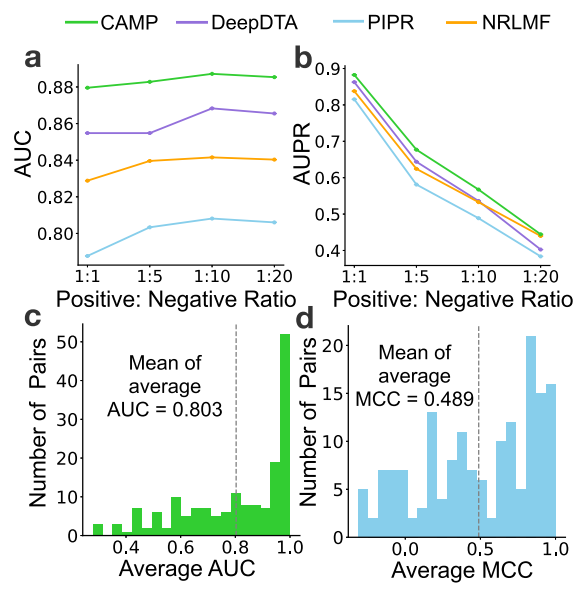
图4. CAMP的鲁棒性测试
CAMP在三个相关任务中的扩展应用
作者进一步研究了CAMP在三个相关任务中的应用潜力,即预测peptide-PBD(蛋白结合域)相互作用预测、结合亲和力评估和多肽的虚拟筛选。作者将CAMP与两种HSM模型进行了比较,即HSM-ID和HSM-D预测peptide-PBD相互作用。在这里,比较了CAMP和HSM模型在预测peptide-PBD相互作用方面的性能。特别是,在HSM论文中使用相同的数据集和8倍交叉验证设置来评估CAMP的性能。图5显示,除PDZ族外,所有结构域族的CAMP均显著优于HSM-ID和HSM-D。
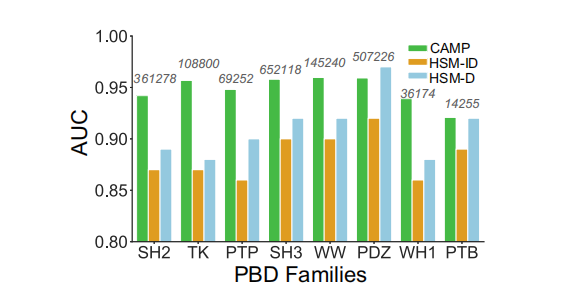
图5. CAMP、HSM-ID和HSM-D跨8个族的模型性能
4
总结
作者开发了一个可多层次预测多肽-蛋白相互作用的深度学习框架(CAMP),包括二元相互作用预测和多肽结合残基预测。方法使用多通道特征提取器分别处理数值特征和分类特征,以避免多源特征的不一致性。此外,作者还提出了四个具有代表性的案例来可视化多肽结合残基鉴定任务的结果。同时,验证了CAMP在peptide-PBD相互作用预测、多肽-蛋白对的结合亲和力评估和多肽的虚拟筛选方面的应用潜力。这些结果表明,CAMP可以提供准确的多肽-蛋白相互作用预测,并为理解多肽结合机制提供有用的见解。
参考文献
Yipin Lei, Shuya Li, Ziyi Liu, Fangping Wan, et al. A deep-learning framework for multi-level peptide–protein interaction prediction. Nature Communications, 2021, 12(1): 5465.
代码 | 数据:
https://github.com/twopin/CAMP
文章来源: drugai.blog.csdn.net,作者:DrugAI,版权归原作者所有,如需转载,请联系作者。
原文链接:drugai.blog.csdn.net/article/details/120559054

- 点赞
- 收藏
- 关注作者
评论(0)