计算化学的深度学习

摘要
人工神经网络的兴衰在计算机科学和计算化学的科学文献中都有详细记载。然而近二十年后,我们现在看到了对深度学习兴趣的复兴,这是一种基于多层神经网络的机器学习算法。在过去的几年里,我们看到了深度学习在许多领域的变革性影响,尤其是在语音识别和计算机视觉领域,在这些领域的大多数专家从业人员现在经常避开之前建立的有利于深度的模型学习模型。在这篇综述中,我们对深层神经网络理论及其独特性质进行了介绍性概述,将它们与化学信息学中使用的传统机器学习算法区分开来。通过概述深度神经网络的各种新兴应用,我们强调它的普遍性和广泛的适用性,以应对该领域的各种挑战,包括定量结构活性关系,虚拟筛选,蛋白质结构预测,量子化学,材料设计和财产预测。在回顾深度神经网络的表现时,我们观察到在不同的研究课题中,针对非神经网络最先进的模型的一致表现优异,而基于深度神经网络的模型通常超出了各自任务的“玻璃天花板”预期。加上用于训练深度神经网络的GPU加速计算的成熟度以及用于训练这些网络的化学数据的指数增长,我们预计深度学习算法将成为计算化学的宝贵工具。
介绍
深度学习是AlphaGo开发过程中使用的关键算法,AlphaGo是由Google开发的Go-playing程序,在2016年击败了顶级人类玩家。 在棋盘游戏中击败人类玩家的计算机程序的发展并不新鲜; IBM的棋牌电脑Deep Blue在二十年前的1996年击败了顶级棋手。不过,值得注意的是Go可以说是世界上最复杂的棋盘游戏之一。 在19 *19板上玩,大约有10170个法律职位可以玩。 与Go的复杂性相比,据估计Lipinski虚拟化学空间可能只包含1060个化合物。
深度学习是一种机器学习算法,与计算化学中各种应用中已经使用的算法不同,从计算机辅助药物设计到材料性质预测。其中一些更高调的成果包括2012年默克活动预测挑战,其中 一个深层的神经网络不仅赢得了比赛,而且超越了默克的内部基线模型,但是在他们的团队中没有一名化学家或生物学家的情况下这样做。 在一个不同的研究团队的不断取得的成功中,深度学习模型在2014年NIH发布的Tox21毒性预测挑战中取得了最高的地位。在最近的这些例子中,深度学习模型在预测活性和毒性方面的异常出色的表现来源于独特的特点,区别于传统机器学习算法的深度学习。
对于那些不熟悉机器学习算法复杂性的人,我们将重点介绍一些主要差异-传统(浅层)机器学习和深度学习之间。机器学习算法最简单的例子就是无所不在的最小二乘线性回归。在线性回归中,模型的基本性质是已知的(在这种情况下是线性的),而输入(也称为模型的特征)彼此是线性独立的。通过变换原始数据(即平方,取对数等)可以将额外的复杂性添加到线性回归中。随着更多这些非线性项被添加,回归模型的表现力增加。这个描述突出了传统(浅)机器学习的三个特点。首先,这些功能由领域专家提供。在一个被称为特征提取和/或工程的过程中,应用了各种变换和逼近,这可以从第一原理出发,或者可能是众所周知的近似,甚至是受过教育的猜测。其次,浅层学习是模板匹配。它不会学习问题的表示,它只会学习如何精确地平衡一组输入要素以产生输出。第三,它的表达能力随着项的数量(即要拟合的参数)而增长,但如果非线性变换选择得不好,它可能需要指数多项。 例如,简单的幂级数展开将需要大量的项(和参数)来适应具有大量振荡的函数。 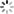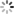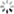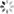
图1. a)具有一个隐藏层的传统前馈人工神经网络(ANN)的示意图。 每个表示为圆圈的神经元接受一系列n个输入值,并将其映射到使用非线性函数的输出,并将隐含层的所有神经元应用于偏置项(即输入为零时的神经网络输出)。 b)深度神经网络(DNN)与人工神经网络的不同之处在于具有多个(n> 3)隐藏层,如示意图所示,为简单起见,这里省略了偏差项
目前开发的绝大多数深度学习算法都是基于人工神经网络的,为了本次审查的目的,我们将专注于深度神经网络。在本综述的前半部分,我们将提供深入学习的简要非技术性介绍,从人工神经网络的基本背景开始,并突出介绍在过去十年中使深度神经网络成为可能的关键技术发展。此外,我们将重点关注深度学习与计算化学中使用的传统机器学习算法的不同之处,以及深度学习正在进行的复兴与20世纪80年代的人工神经网络模型如何不同,后者可能被视为其“父”算法。在接下来的半年回顾中,我们将对计算化学领域的深度学习应用的最新发展进行调查,我们将根据现有的机器学习模型来检验其性能,以及为该领域做出贡献的未来前景。这篇评论主要是为了作为计算化学家的介绍入口,这些计算化学家希望从应用的角度探索或整合深度学习模型在他们的研究中,并且将提供对现有文献综述的更多参考资料,以涵盖更深层次的技术方面学习神经网络结构和优化。
深度学习101
人工神经网络(ANN)是大多数深度学习算法的基础,它是一类受生物神经网络启发的机器学习算法,用于通过将大量输入转换为目标输出来估计或近似函数。 1a)中,人工神经网络由一系列层构成,每层包含许多“神经元”。每个神经元接受来自前一层的输入值,并将其映射到非线性函数上。该函数的输出用作ANN中下一层的输入,直到它到达最后一层,输出对应于要预测的目标。此外,可调参数,每个神经元函数的“权重”(或系数)在构建该模型时进行调整,以最小化预测值的误差,这一过程称为“训练”神经网络。形象地说,人工神经网络中的这些神经元的集合模仿神经元在生物系统中的工作方式,因此其名称为人工神经网络。
在反向传播过程中,使用称为梯度下降的算法来在生成相应输出时查找由各个神经元引起的误差表面中的最小值。 从概念上讲,梯度下降与经典分子动力学模拟中使用的最速下降算法没有区别。 主要区别在于迭代地最小化能量函数并更新每个步骤的原子坐标,迭代地使ANN的目标输出的误差函数最小化并且每步更新神经元的权重,在ANN文献中,这也被称为“迭代”。 训练集中的数据可以迭代多次,并将数据完整地传递为“epoch”。
反向传播的一个关键问题是随着信号通过每个隐藏层,误差信号逐渐变得更加分散。 这是因为信号发生了在模型更深处,越来越多的神经元和权重与给定的误差相关联。 直到最近,这使得很难有效地训练许多层; 超过几层的任何东西都需要很长时间才能收敛到过度拟合的高可能性,特别是对于最接近输出的层。 此外,非线性变换函数(如S形)具有有限的动态范围,因此误差信号在通过多个层时趋于衰减,这通常称为“消失梯度问题”。
自1986年以来,已经开发了几种关键算法,包括无监督预训练,纠正线性函数和dropout,以改进人工神经网络的训练过程,解决消失梯度问题,并减少过拟合人工神经网络特别容易受到影响。也许是训练深度神经网络(DNN)的最大障碍,是渐近问题的消失,因为它实际上限制了神经网络的深度。预训练,由Hinton等人发现。在2006年是一种快速,贪婪的算法,它使用无监督的分层方法一次训练DNN一层。预训练阶段完成后,将使用更细微的微调过程(例如随机梯度下降)来训练模型。使用预训练方法,模型在反向传播开始之前就已经学会了这些特征,从而减轻了消失梯度问题。 2011年出现了另一种解决方案,Bengio及其同事证明整流线性激活(ReLU)函数完全避开了消失梯度问题。 ReLU的一阶导数恰好为1或0,通常可以确保误差信号能够反向传播而不会消失或爆炸。 (图2)。
图2. a)S形和 b)整流线性(ReLU)函数(蓝色)及其相应的一阶导数(红色)的图。 与sigmoidal函数不同,其中的导数根据x的值而变化,在ReLU函数中,一阶导数为0或1。[Color figure can be viewed at wileyonlinelibrary. com]
总结了人工神经网络及其相关算法的主要发展情况后,我们注意到它并不全面。除了迄今为止讨论的传统的前馈DNN(图1b)之外,更近期的发展包括替代架构,特别是卷积神经网络(图3a),递归神经网络(图3b),和自动编码器(图3c),这些在计算机视觉和自然语言处理应用中都非常成功。关于各种DNN体系结构的技术讨论,虽然对深入学习文献有深刻理解,但超出了本文的讨论范围,因此,我们向读者推荐以下出版物总结这一研究课题。现在,应该明显看到,ANN本身不是一项新发明。事实上,人工神经网络的数学算法是由McCulloch和Pitts在1943年开发的,实际上可训练的人工神经网络可追溯到1986年,与Rumelhart,Hinton和Williams等人发明神经网络的后向传播一致。更深层次的神经网络超越了几个隐藏层(图1b),只有在最近几年更新的算法发展才能实现。因此,DNN不仅仅是上个世纪人工神经网络的重塑品牌,它又如何更好比已经成功用于各种化学信息学应用的传统机器学习算法?
图3. a)卷积神经网络(CNN)的示意图。 CNN的设计明确假定输入是以图像数据的形式。 每个卷积层提取和保存空间信息并学习一个表示,然后典型地将其传递到输出层之前的传统完全连接的前馈神经网络。 b)循环神经网络(RNN)的示意图。 最简单实现的RNN是对标准前馈神经网络的修改,其中隐藏层中的每个神经元从模型的前一次迭代的输出接收附加输入,表示为“t-1”圆形箭头。 c)自编码器的示意图,它是用于无监督学习的神经网络。 在自动编码器中,目标是学习输入层的标识函数,并且在该过程中,隐藏层中原始数据的压缩表示被学习。
数十年的化学研究已经导致了几千种分子描述符的发展,这些描述符描述了可以想到的任何化合物的一系列性质。因此,分子描述符用作使用化学知识和直觉(即领域专业知识)构建的特征,其可用于传统的机器学习模型,其已经在计算化学应用中取得了合理的成功。传统的机器学习算法(如线性回归和决策树)非常直观,可以创建人类可以理解的简单模型。尽管如此,随着我们对具有非线性关系的更复杂属性(通常是与生物过程和材料工程相关的属性)的预测进展,通常需要依赖更复杂且不太透明的算法,如支持向量机(SVM)和随机森林 (RF)达到可接受的预测准确度。乍一看,深度学习算法属于后一类,但它有一个主要区别。与SVM和RF不同,DNN转换输入并将其重构为跨隐藏层的神经元的分布式表示。通过适当的训练方法,系统隐藏层中的神经元将学习不同的特征;这被称为自动特征提取。由于每个隐藏层都成为系统下一层的输入,并且可以沿途应用非线性转换,因此它会创建一个逐渐“学习”越来越抽象,层次和深度特征的模型。
自动特征提取是一个不需要领域知识的过程,因此是深度学习算法最重要的优点之一。这与传统的机器学习算法不同,在这种算法中,模型必须用基于化学知识和直觉的“正确”特征仔细构建,以便其执行和推广。正因为如此,深度学习已成为语音识别和计算机视觉中的主流算法。 ImageNet是用于图像分类的各种算法的年度评估和竞争。在进行深度学习之前,所采用的最先进的模型徘徊在25-30%的错误率,这与理想的匹配5.1%经过训练的人为错误率的理想目标相距甚远。 2012年,Hinton及其同事首次向这个社区介绍了深度学习算法,他们的基于DNN的模型实现了16.4%的错误率。这是当时计算机视觉中已建立的模型的显着改进,而基于传统机器学习算法的次高性能模型仅实现了26.2%的错误率。随后在基于DNN的模型中的改进最终实现了5.0%以下的错误率,超过了2015年的人类表现(图4),这是深度学习引入计算机视觉领域后仅3年对于这些领域的从业人员来说,深度学习和自动特征提取能力的影响已经发生了变革,不仅超越了该领域的“玻璃天花板”期望的超越能力,而且实现它的时间也显着缩短。近年来,深度学习在计算机科学领域以外的其他学科中也有所表现,包括高能粒子物理和生物信息学。
图4. 年度ImageNet竞赛中表现最好的图像分类算法的历史错误率。已建立的计算机视觉模型停滞在25-30%。 2012年引入深度学习使得图像分类的显着改善达到了~15%,到2015年实现了图像分类的人类精确度(~5%)。
还没有讨论过的深度学习的一个同样重要的方面是多年来非算法发展的作用。具体来说,由于上个世纪缺乏“大数据”的可行性和GPU硬件的技术进步,导致了DNN的出现不同于上个世纪的ANN。 2012年的开创性工作被广泛认为是推动深入学习受到关注的文章,是Hinton的AlexNet论文。虽然算法的发展,尤其是dropout促成了它的成功,但可以获得包含120万张图像的更大的数据集到图像中使用的10,000幅图像的数据集,也对其成功起到了关键作用。随着更深更大的神经网络的发展,训练时间通常可能延长至数天或数周。然而,就像计算化学领域如何从GPU加速计算的兴起中受益一样,该技术也减轻了DNN的训练速度问题。
在更实际的考虑中,用于在GPU上训练神经网络的开源代码和文档的可用性也可以说是近年来深度学习快速扩散的另一个原因,包括其对学术研究的影响,这可以通过自2010年以来深度学习相关出版物指数级增长看出来(图5a)。就像现代大多数计算化学家不再编写他们自己的代码来执行分子动力学模拟或运行量子化学计算一样,而是依赖于已建立的软件包,深度学习研究团队也已达到类似的成熟度水平,目前用于训练神经网络的主要软件包,包括Torch,Theano,Caffe和Tensorflow。Torch也许这四者中最古老的一个,其作为一种机器学习科学计算框架于2002年首次在纽约大学发布,自那时起,深度学习库被添加。 Theano是2008年由Benjio及其同事在蒙特利尔大学发布的第一个专门开发的深度学习框架,之后,该框架已发展成为一个由250多名贡献者组成的社区团队。 2014年由伯克利视觉与学习中心开发的Caffe的发布紧随其后。最近,由谷歌开发的Tensorflow于2015年下半年发布,可以说在深度学习社区中的吸引力激增,从谷歌搜索排名的高峰(图5b)可以看出,以及它的Github已经分别出演了33,000次和14,000次,尽管它仅仅在一年多的时间内发布了一次。此外,2015年发布的Keras等API大大简化了神经网络的构建和培训,显着降低了新的深度学习从业者的入门门槛。
图5. 来自ISI索引的 a)深度学习出版物的增长情况,以及 b)2010年以来主要深度学习软件包的年度Google趋势得分。
毫无疑问,计算机科学领域一直是从互联网上获得的可挖掘数据激增的主要援助者(图6a),毫不奇怪,深度学习的影响力也是最大的。在化学领域,我们也看到了可公开访问的数据库(如Protein Data Bank(图6b)和PubChem(图6c))中数据的相应增长,其中更多的数据来源于最新的高通量组学技术[53]。正是由于这些原因,我们对计算化学领域开始经历同样的事件汇合感到乐观,这将极大地促进我们领域的深度学习应用。我们可以利用计算机科学领域的算法突破,化学数据的日益增加的可用性,以及现在成熟的GPU加速计算技术。[ 图6d; GPU计算能力数据点来自所报道的双精度(2010),M2090(2011),K20(2012),K40(2013),K80(2014),P100(2015)计算NVIDIA Tesla系列GPU的计算能力。]
图6. a)生成的全球数据,b)保存在蛋白质数据库中的结构数量,c)保存在Pub-Chem中的化合物数量,以及d)用于科学计算的GPU计算能力(GPU计算功率数据点 (2010年),M2090(2011年),K20(2012年),K40(2013年),K80(2014年),P100(2012年),P100(2011年),NVIDIA Tesla系列GPU的双精度计算能力, 2015)),在它们向上的轨道上都有类似的相似之处
计算机辅助药物设计
在计算机辅助药物设计中,传统的机器学习算法在化学信息学领域有着悠久的历史,特别是它们对量化结构活性关系(QSAR)应用的贡献。在QSAR中,要预测的输出通常是化合物的生物活性。通常使用回归模型,并且输入数据是分子描述符,其是根据化学领域知识设计的分子的预先计算的物理化学性质。 QSAR应用的早期工作使用线性回归模型,但这些模型很快被贝叶斯神经网络所取代,随后是RFs和SVMs 。该领域的从业者历来喜欢允许可变选择的模型,以便知情的化学家可以确定选定的特征是否有意义。此外,也允许评估输出预测不确定性的模型。 QSAR领域非常广泛,我们向读者推荐以下关于历史关键技术发展的评论列表[57-60]。为了进行本次审查,我们将讨论的范围限制在基于DNN的QSAR模型的性能和适当比较传统的机器学习模型。
深入学习QSAR的第一次尝试是2012年的默克挑战。 在这个公开的挑战中,团队提供了化合物的预计算分子描述符及其相应的实验测量活动,共计15个药物靶标。 提交的模型评估了他们根据没有发布给参与者的测试集预测活动的能力。 获胜组使用DNN模型,由达尔领导,他是Hinton研究团队的一员。值得注意的是,应该强调的是,该团队没有受过正式训练的计算化学家。 他们来自计算机科学系。
2014年,Dahl等人根据Merck挑战中使用的算法提交了预发布论文,探讨了多任务神经网络在QSAR应用中的有效性。在这项工作中,作者使用了一个多任务DNN模型。在这里,“多任务”指的是一种模型,它不仅预测单个感兴趣的输出,而且预测同时多个输出,在他们的情况下是19次分析的结果。使用的数据集来自PubChem,包含超过100,000个数据点。分子描述符每个分子总共3764个描述符使用Dragon生成,并且它们被用作DNN的输入特征。在与其他传统机器学习算法(例如梯度推进决策树和逻辑回归)相比的准确性性能基准中,基于DNN的模型在19个化验预测中的14个中的表现优于统计上显着的余量,并且在性能剩余的5个化验预测。另外,作者指出了多任务神经网络的优点,特别是它为多任务开发了一个共享的,学习的特征提取流水线。这意味着不仅可以学习更一般的特征产生更好的模型,而且多任务DNN中的权重也受到更多数据案例的约束,从而分享统计强度。最后,该研究的一个有趣的观察结果是DNN如何处理数以千计的相关输入特征,这与Winkler在2002年强调的传统QSAR智慧相反,尽管我们注意到Winkler当时发表的观察结果是在开发DNN之前。在达尔的工作中,作者观察到,将输入特征减半不会导致性能下降。
Merck于2015年发表的一项随后研究综合分析了DNN的培训,并将其性能与该领域当前使用的基于RF的模型的性能进行了比较,并将其扩展到Merck挑战数据集。作者总结说,DNNs可以作为一种实用的QSAR方法被采用,并且在大多数情况下容易胜过射频模型。就实际采用而言,作者强调了DNN利用的GPU硬件的巨大进步,以及与传统机器学习模型使用的传统CPU集群相反的部署GPU资源的经济成本优势。还研究了与训练深度神经网络相关的关键问题,特别是可调参数的数量。作者发现,大多数单任务问题可以在具有两个隐藏层的架构上运行,每层仅使用500-1000个神经元,并且使用75个训练时期。更复杂的体系结构和/或更长的培训时间可以提高模型精确度的回报,但增量递减。尽管在上文总结的Merck挑战和相关研究中,DNN总体上表现良好,但研究界的一些从业人员对此结果持怀疑态度。常见的问题包括样本量小,并且面对模型复杂性的增加,预测准确性的渐进式改进难以证明其合理性。
2014年,Hochreiter及其同事在神经信息处理系统(NIPS)会议上发表了一篇同行评议的论文,讨论如何将多任务DNN应用于QSAR应用于一个更大的数据集。在这项研究中,作者策划了整个ChEMBL数据库,该数据库比原始Merck挑战数据集大两个数量级。该数据集包括743,336种化合物,约1300万种化学特征和5069种药物靶标。有趣的是,作者没有使用明确计算的分子描述符作为输入数据,而是使用了ECFP4指纹。作者对1230个目标的DNN模型的准确性进行了基准测试,并将其与传统的机器学习模型(包括SVM,逻辑回归等)进行了比较。应该指出,在2014年Dahl的论文中,梯度提升的决策树与DNN差不多,并未包含在本研究中。然而,事实表明,DNNs的表现优于所有测试的模型,其中还包括两家商业解决方案和三家制药公司目前实施的解决方案(图7)。虽然大多数传统机器学习算法的准确度范围为0.7至0.8 AUC,但DNN达到了0.83的AUC。在更好的表现模型(AUC> 0.8)中,DNNs也具有最不严重的异常值,这些作者假设是由于DNN的共享隐藏表示使得它能够预测单独检查时难以解决的任务。与达尔2014年的研究一致,多任务DNN的使用赋予了两个优点:(i)允许多标记信息,因此利用任务之间的关系和(ii)允许在预测任务之间共享隐藏的单位表示。这项研究的作者指出,第二个优点对于一些仅有少量测量数据的药物靶标特别重要,因此表明单个目标的预测可能无法构建有效的表示。使用多任务DNN可以部分缓解这个问题,因为它可以利用跨不同任务学习的表示,并且可以用较少的训练示例提高任务的性能。此外,DNN提供了化合物的分层表示,其中更高层次表示更复杂的概念,这些概念可能会超出训练集数据的潜在更多转移。
图7.深度神经网络与几种传统机器学习算法的性能准确性(就AUC度量而言),包括:支持向量机(SVM),逻辑回归(LR),k-最近邻(k-NN)和商业实现 解决方案(Pipeline Pilot Bayesian分类器,分别基于Parzen-Rosenblatt KDE的方法和相似性集成方法),用于从ChEMBL获得的策展数据库的活动预测。
Pande group和Google在2015年向arxiv提交了一项类似的大规模研究。在这项研究中,确定了大约200个药物靶点,但包括了更多的数据点(4000万)。与早期的NIPS论文不同,Pande及其同事将调查重点放在了DNN多任务学习的有效性上,而不是DNN模型本身的性能。作者策划了一个数据库,该数据库由多个公开数据源组成,包括来自PubChem数据库的PCBA ,来自17个具有挑战性的虚拟筛选数据集的MUV ,DUD-E组[和Tox21数据集。与Hochreiter及其合作者一样,这些分子使用ECFP指纹进行了修饰,并且没有计算明确的分子描述符。主要研究结果中,一直观察到多任务绩效改善,但其他数据或额外任务在改善绩效方面是否有较大影响并不明显。作者还观察到训练集中未包含的任务的转移能力有限,但效果并不普遍,并且在成功运行时需要大量数据,这部分强化了Hochreiter和Dahl提出的多任务学习优势的主张。奇怪的是,从一个数据集到另一个数据集,多任务改进程度各不相同,并且没有提供令人满意的解释。尽管如此,多任务DNNs与逻辑回归和RF等传统机器学习模型的一致性能显着(图8),其中AUC的性能提升范围从0.02到0.09。
图8.当使用多任务深度神经网络(MT-DNN)时,与逻辑回归(LR)、随机森林和单任务神经网络(ST-NN)相比,在3个不同数据库(PCBA,MUV,Tox21)中观察到的准确性(以AUC度量指标) (RF)。
迄今为止,至少有四个DNN报告应用于QSAR,一致的观察结果表明深度学习优于传统的机器学习对手。然而,所有研究都主要集中在生物活性预测上。从概念上讲,DNN在预测其他感兴趣的性质(其可能包括ADMET性质)以及在计算机辅助药物设计的其他部分中的应用(例如在虚拟筛选中)应该具有类似的性能。
药物性肝损伤(DILI)是过去五十年中安全性相关药物撤药的最常见原因。 DILI的机制是复杂和多样的,在人类中引起DILI的药物不易通过常规方法探测,使DILI的毒理学研究变得困难。徐等人最近的一项研究表明,使用DNN预测DILI毒性。作者使用了Mold和PaDEL计算的显式分子描述符,以及Lusci等开发的URGNN分子构建编码方法作为DNN的输入数据。该模型接受了475种药物的培训,其中198种药物的外部测试集合,使用DNN的最佳模型的准确率为86.9%,灵敏度为82.5%,特异性为92.9%,AUC为0.955。相比之下,传统模型的绝对性能指标降低了10-20%。有趣的是,使用来自URGNN分子结构编码方法的输入,作者创建了性能最高的模型(AUC 0.955),优于经类似训练的DNN,其使用来自Mold(AUC 0.931)和PaDEL(AUC 0.895)的计算分子描述符。这表明,一种好的分子编码方法如UGRNN可能更有效地为DNN提供必要的特征,因为深度学习算法具有自动提取必要特征的能力,并且这种能力可能相当于或甚至可能优于域通过开发明确的分子描述符来进行专家特征工程。
Swamidass及其同事于2015年发布了DNN建模毒性的另一个应用。药物毒性的一种常见机制源自与蛋白质共价结合的亲电子反应性代谢物。环氧化物是这种性质的官能团,其通常由药物分子的细胞色素P450代谢形成,其作用于芳族或双键。 Swamidass和同事的结果特别有特色,因为他们开发了DNN模型来预测经历环氧化的分子的特定位置,即其环氧化(SOE)位点。这项工作的基础是早期的模型Xenosite,一种基于ANN的小分子P450代谢模型,尽管它是一个浅层网络,但它已经超越了基于SVM的模型的精确度高达5%。随后通过研究使用不同类型的分子指纹对P450代谢建模的影响进一步改进,他们发现使用不同指纹类型的共识模型可以获得进一步的准确性增加和预测的相关姊妹模型葡萄糖醛酸化代谢的位点。在他们最近关于环氧化物基毒性预测的工作中,Swamidass及其同事设计了一种4层DNN结构,并在702环氧化反应数据库上训练了模型,并鉴定出94.9%AUC性能的SOE,并将其分离(即分类)具有79.3%AUC的环氧化和非环氧化分子。此外,在环氧化分子内,该模型能够通过将芳香族或双键SOEs与所有其他芳族或双键分别具有92.5%和95.1%的AUC分离来提供原子级精确信息。这使得DNN模型成为文献中的第一个机理模型,它不仅预测候选药物的反应性环氧化物的形成,而且准确地识别分子中的特定环氧键。 Swamidass及其合作者使用类似的DNN模型模拟了小分子对软亲核试剂如谷胱甘肽(GSH)的反应性位点。通过仅对定性反应性数据进行培训,他们能够构建基于DNN的模型,其以90.8%的准确度识别反应性分子内的反应性位点,并分离具有80.6%准确度的反应性和非反应性分子。此外,该模型的预测与更具化学多样性的外部数据集中的定量GSH反应性测量结果相关性很好,这表明该模型在更大面积的化学空间范围内具有普遍性。随后的出版物扩大了该模型的范围,以涵盖对GSH,氰化物,蛋白质和DNA的反应性。由此产生的模型对DNA进行交叉验证的AUC性能为89.8%,对于蛋白质为94.4%,并且将来自非反应性分子的亲电子反应性分子与DNA和蛋白质分开,交叉验证的AUC性能分别为78.7%和79.8% 。此外,该模型的表现也显着优于用QM方法计算的反应性指数。由于药物毒性通常是由亲电子反应性代谢产物引起的,因此有助于识别位点反应性研究的模型(迄今为止在文献中显着缺失)可以用于构建基于机制的分子毒性预测。
2016年Hochreiter集团最近还发布了一项关于化学毒性的大规模研究。在这项工作中,作者报告了2014年NIH发布的针对Tox21数据挑战的DNN模型的应用。该数据库由12,000种环境化学品和药物组成,并且它们在12种不同测定中的相应测量被设计用于测量多种毒性效应。毫不奇怪,由Hochreiter及其同事开发的DeepTox模型在提交给Tox21挑战的所有方法中性能最高。对其模型的进一步分析表明,使用多任务DNN模型导致在12个化验预测中的10个中与单任务模型相比有一致的优势。传统机器学习算法(包括SVM,RF和Elastic Net)的其他基准也证明了DNN在15个案例中有10个胜出。最后,尽管最初的DeepTox模型使用NIH提供的分子描述符在Tox21挑战中,作者还表明,仅使用ECFP4指纹作为输入数据开发的类似训练的DNN模型具有与那些在显式分子描述符上训练的类似的性能,其类似到Xu等人的观察。在他们的DILI毒性模型中。有趣的是,在可视化这些DNN的第一个隐藏层时,作者观察到该层中99%的神经元与至少一个已知毒性特征具有显着关联,这表明深度学习可能支持发现新的化学知识其隐藏层。
根据QSAR和毒性预测的进展,深度学习算法也开始对计算机辅助药物设计的其他方面产生影响。 2013年,Baldi和同事报告使用DNN模型预测分子溶解度[78]。 Pande及其同事还向arxiv提交了此方向最近的研究进展,他们开发了一个多任务DNN模型,用于预测不仅溶解度,而且预测ADMET性质的整个范围。深度学习在虚拟筛选方面也可能具有未来作为现有对接方法的可行替代或补充。 2016年,AtomNet提交了一份arxiv论文,该公司开发了一种DNN模型来分类停靠在蛋白质结合口袋中的小分子的活性[87]。值得注意的是,AtomNet DNN模型能够达到0.7到0.9之间的AUC指标,具体取决于所使用的测试集,它明显优于传统的对接方法,特别是Smina,[88] AutoDock Vina [89]分叉0.1到0.2。 [87]关于与计算生物学更紧密结合的应用中的深度学习的其他近期发展,我们向读者推荐以下关于该研究课题的评论。[90]
计算结构生物学
当蛋白质序列折叠成其三维结构时,预测蛋白质序列的任何两个残基的空间接近性称为蛋白质接触预测。对顺序不同残基之间接触的预测因此对其3D结构施加强约束,使其对从头蛋白质结构预测或工程特别有用。虽然使用基于物理学的模拟方法,如长时间尺度分子动力学[91,92]可用于从头算蛋白质结构预测,但计算需求是艰巨的。 Wolynes,Onuchic等人开发的互补方法,如基于知识的物理方法也是一种选择[93,94],虽然它们的计算成本较低,但仍然要求足够高,以至于不能用于大规模研究。因此,机器学习方法是可行的替代方案,包括基于人工神经网络,[95-97] SVM,[27]和隐马尔可夫模型[98]的方法。其他方法包括基于模板的方法,这些方法使用同源性或线程方法来识别结构相似的模板,以推断蛋白质接触预测。[99,100]接触预测因子的这些不同模型的评估是蛋白质关键评估的亮点之一结构预测(CASP)的挑战始于1996年。尽管多年来有所改进,但远程接触预测历史上已经达到了精确度低于30%的玻璃上限。计算蛋白质结构预测的关键历史发展是大量的,我们引用感兴趣的读者对这个主题的现有评论。[101-104]为了这次审查的目的,我们将讨论范围限制在最近的DNN-基于模型的模型,以及它们如何对突破玻璃天花板的历史预期至关重要。
2012年,Baldi和同事们开发了多阶段机器学习方法CMAPpro,将接触预测的准确性提高到36%[105]。 CMAPpro在早期模型中实现了三项具体的改进。首先是使用二维递归神经网络来预测二级结构元素之间的粗糙接触和取向。此外,一种新型的基于能量的神经网络方法被用来改进来自第一网络的预测,并用于预测残留物接触概率。最后,DNN架构被用来通过整合空间和时间信息来调整所有残差 - 接触概率的预测。 CMAPpro接受了来自ASTRAL数据库的2356个成员训练集的训练。为了交叉验证的目的,该组被分割成属于不同SCOP折叠的10个不相交的组,这意味着无论是训练还是验证都设置共享序列或结构相似性。然后对所产生的模型性能进行测试,对照在ASTRAL数据库版本1.73和1.75之间报告的364个成员的新蛋白质折叠测试集。将CMAPpro性能与多级机器学习模型的几种排列进行比较,包括单隐层神经网络(NN),单隐层神经网络,其利用由2D递归神经生成的粗接触/定向和对准预测器网络和基于能量的神经网络(NN1CA)以及深度神经网络,但没有CA特征(DNN)。基于相对性能,深度网络架构和CA特性都需要达到36%的准确度; DNN和NN1CA各占32%,而代表先前技术水平的NN仅达到26%的精度。
Eickholt和Cheng在2012年也报道了DNN用于蛋白质接触预测的不同实施。[107]在他们的算法DNCON中,它将深度学习与用于开发集合预测器的提升技术相结合。使用来自蛋白质数据库的1426个成员数据集来训练DNCON,训练(1230个成员)和确认(196个成员)组之间随机分配。显式设计的特征被用作DNN的输入。具体而言,使用了三类特征:(i)来自以所讨论的残基对为中心的两个窗口的特征(例如,预测的二级结构和溶剂可及性,来自PSSM的信息和可能性以及Acthley因子等),(ii) )成对特征(例如Levitt的接触电位,Jernigan的配对潜能等)和(iii)全局特征(例如,蛋白质长度,预测的暴露的α螺旋和β片层残基的百分比等)。使用这些工程功能,DNN模型的任务是预测是否有特定的残基对接触。此外,分类器的增强集合是通过使用从训练集中获得的较大池中的90,000个长程残留 - 残留对的样本训练几个不同的DNN而创建的。在评估其性能时,DNCON的交叉验证准确率为34.1%。该模型的性能可转移性在其性能基准中与CASP9,[108] ProC_S3,[28]和SVMcon [27]中两个分别基于RF和SVM算法的最佳预测指标进行了对比。在该评估中,每个软件都使用了相应的测试集。虽然改进不如Baldi及其同事报道的那么戏剧化,但DNCON的性能比当时最先进的算法好3%; ProC_S3(32.6%比29.7%)和SVMcon(32.9%比28.5%)。
基于DNN的蛋白质接触预测模型值得注意,因为它使社区能够突破前几年不可能实现的30%的准确性障碍。除蛋白质接触预测外,DNNs也已成功应用于仅从序列数据预测各种蛋白质角度,二面角和二级结构。使用DNNs,Zhou,Yang和同事发表了一系列基于序列的钙基角度和扭转预测[109-111]。与蛋白质接触预测不同,骨干扭转可以更好地限制从头蛋白质结构预测和其他建模目的。[112]在这些基于DNN的模型的开发中,Zhou,Yang及其合作者使用了4590个成员的训练集和从蛋白质序列剔除服务器PISCES获得的1199个独立测试集[113]。输入数据特别包括从PSI-BLAST产生的位置特异性评分矩阵[114,115]获得的工程特征以及与残基特性有关的几个其他物理化学特性,包括空间特性,疏水性,体积,极化率,等电点,螺旋概率等。[116]
在SPINE-X算法的开发中,使用DNN直接预测二级结构,残余溶剂可接近表面积(ASA)和u和扭转角[110]。开发了一个六步机器学习体系结构,其中ASA等输出被用作其他要预测的属性(如扭转)的后续输入。基于模型在独立测试集上的性能评估,它分别获得了u和w两面体的平均绝对误差228和338。独立数据集的二级结构预测精度范围为81.3%至82.3%,考虑到序列数据的二级结构预测领域近十年来停滞在近80%的准确度范围内,这一成就值得注意,其中一些利用传统机器学习算法。[117]以类似的方式,对于稍后开发的SPIDER算法,DNN被用于直接预测Ca角(h)和扭转(s)。根据对模型性能的评估,它分别获得了h和s的平均绝对误差98和348,作者观察到该模型的误差从螺旋残留物增加到片残留物到残余物,随后出现非结构化趋势。使用这些预测的角度和扭转作为限制,作者能够在预测结构和天然结构之间对蛋白质的三维结构进行建模,平均RMSD为1.9A。随后,SPINE-X和SPIDER算法作为并行多步算法重新训练,同时预测以下性质:二级结构,ASA,u,w,h和s。这导致二级结构整体精度略有提高2%,并且角度/扭矩MAE减少1-38,同时保持相同的ASA性能水平。
除了蛋白质结构建模之外,深度学习也被用来根据序列数据预测其他感兴趣的特性。例如,最近报道预测DNA和RNA结合蛋白的序列特异性[118,119]。在Frey及其合作者的开创性研究中[118],DeepBind算法用于预测DNA和RNA结合蛋白的序列特异性。使用12TB的序列数据,跨越数千个公共PBM,RNAcompete,ChIP-seq和HT-SELEX实验,将原始数据用作DNN算法的输入以计算预测结合分数。 DeepBind表征DNA结合蛋白特异性的能力在来自Weirauch等人修订的DREAM5 TFDNA基序识别挑战的PBM数据中得到证实[120]值得注意的是,DeepBind基于Pearson相关性和AUC指标优于所有现有的26种算法,并在DREAM5提交的15个小组中排名第一。有趣的是,他们的结果还表明,体外数据训练的模型在体内评分数据中运行良好,表明DNN已经捕获了核酸结合本身的一部分性质。
随着在其他领域重复出现深度学习优于传统机器学习算法[18,32-35]以及计算机辅助药物设计本身[62,67,69],DNN在推动“玻璃”天花板“蛋白质接触预测和二级结构预测的界限应该不令人意外。本次审查中显然缺乏的是深度学习在RNA结构预测和建模中的应用,据我们所知尽管尚未报道。与蛋白质数据库相比,RNA上的可用结构数据更小。此外,大多数RNA结构数据不是结晶学的,而是基于核磁共振的,由于NMR结构本身是用基于物理学的力场对抗实验有界的约束来近似解决的事实,其自身受到较高的不确定性[121]。尽管如此,研究深度学习如何使RNA建模社区受益将会很有趣。
最后,与计算机辅助药物设计相比,在计算结构生物学应用中使用深度学习的一个有趣的对比是对工程特征的独占使用,并且在一些情况下,多级机器学习算法本身的体系结构的工程设计。 虽然计算机辅助药物设计领域的发现是初步的,但有一些迹象表明,明确设计的特征不一定能更好地对抗化学指纹,这可能需要较少的化学领域知识来构建。 尽管我们承认蛋白质比小分子复杂得多,但确定使用仅包含基本结构和连接性信息的输入数据的DNN模型的性能(没有任何特别设计的特征)是否能够准确预测诸如 蛋白质二级结构和远距离接触。
量子化学
使用机器学习补充或取代传统的量子力学(QM)计算已在过去几年出现。在本节中,我们将研究一些机器学习应用到量子化学,并检查类似的基于DNN的模型的相对性能。 2012年,von Lilienfeld和同事开发了一种基于非线性统计回归的机器学习算法,以预测有机分子的雾化能量[29]。该模型使用分子生成数据库(GDB)的7000个成员子集,该数据库是109个稳定且可合成处理的有机化合物库。用于训练的目标数据是使用PBE0杂种功能计算的7000种化合物的原子化能。没有明确的分子描述符被用作输入数据,相反,只有笛卡尔坐标和核电荷用于“库仑”矩阵表示。可以说,没有明确设计的特征,输入数据中的这种表示与传统分子建模方法中使用的分子指纹所提供的表达水平相同。 von Lilienfeld及其同事使用的化合物只有1000种,平均绝对误差(MAE)准确度为14.9 kcal / mol。进一步的外部6000化合物验证组测试产生了15.3 kcal / mol的相似准确度,证明了该模型在“同类”化合物中的可转移性。这项工作特别具有开创性意义的是合理概括QM计算能量的能力,平均绝对误差为15千卡/摩尔,根本没有在机器学习算法中实现薛定谔方程。更重要的是,考虑到这项工作使用了缺乏DNN优势的传统机器学习算法,并且基于DNN的历史性能,这表明基于DNN的模型应该表现得更好。
Hansen等人随后的出版物研究了许多已建立的机器学习算法,以及分子表征对von Lilienfeld工作中使用的相同数据集上原子化能量预测性能的影响[122]。主要研究结果之一是使用“库仑矩阵”的随机变体大大提高了雾化能量的准确度,以实现低至3.0千卡/摩尔的MAE。[122]除了作为分子的逆原子 - 距离矩阵表示之外,随机变体是独特的并且保持关于分子翻译和旋转的不变性。这种改进的表示增加了“副作用”,因为它是高维度的并且考虑到原子的多重索引,所以它是最富有的发展。作者发现,在所有测试的机器学习算法中,通过信息对各种表示进行排序确实会产生相应较低的准确性[122],这强调了QM应用程序中良好数据表示的重要性。公平地说,还应该指出的是,作者确实对人工神经网络进行了基准测试,虽然它们的MAE为3.5kcal / mol,但表现出令人满意的效果,但并没有比非线性回归法的3.0kcal / mol MAE好得多。尽管如此,我们强调所用的神经网络“浅”,有几层,再加上缺少更大的数据集,并不代表真正的DNN实现。本文的一个特别有启发性的猜想是通过外推性能(MAE误差)相对于所使用的数据集的大小,作者得出结论,3千卡/摩尔可能是无论机器学习如何可以实现的“基线”误差算法。[122]
2013年,冯·利林菲尔德报道了第一个多任务DNN模型的应用,该模型不仅可以预测原子化能,还可以预测其他几种电子基态和激发态性质。在这项工作中,他们试图利用多任务学习的优势,通过预测几种电子属性并潜在地捕捉看似无关的属性和理论水平之间的相关性。数据用“库伦矩阵”的随机变量表示。[122]目标数据是使用几种不同的理论水平计算的原子化能量,静态极化率,前沿轨道特征值HOMO和LUMO,电离势和电子亲和力,例如PBE0,ZINDO,GW和SCS。原子化能量保持了0.16eV(〜3.6kcal / mol)的相似MAE精度,并且对于其他能量预测(包括HOMO,LUMO,以及其他)的MAE的准确度为0.11至0.17eV(≥2.5至3.9kcal / mol)电离电位和电子亲和力[123]。此外,这种精确度与用于构建训练集的质量管理计算中使用的相应理论水平的误差相似。
虽然使用机器学习算法替代QM计算是诱人的,但另一种更“第一原理基础”的方法是使用机器学习算法来补充现有的QM算法。正如von Lilienfeld及其同事在2015年首次报道的那样,他们展示了Dlearning方法,即机器学习“修正术语”被开发出来。在该研究中,作者使用DFT计算的特性,并能够使用D学习校正项预测G4MP2理论水平的相应数量。这种复合式QM / ML方法结合了近似而快速的传统QM近似与现代大数据量化的质量管理估算,这些估算在化学空间上进行了昂贵且准确的结果培训。但是,我们注意到,这种方法迄今仅用传统的机器学习算法进行了演示。如果使用我们在众多实例中观察到的多任务DNN进行性能提升适用于此示例,则基于DNN的方法可能会产生出色的结果,但尚未在文献中报告。
据我们所知,量子化学应用中DNN的例子似乎表明,与计算机辅助药物设计和计算结构生物学相比,它处于发展的早期阶段。 从文献中,我们知道传统的机器学习模型已经用于其他质量管理应用,例如建模电子量子传输,用于精确半经验量子化学计算的学习参数[126]等等。 另外,QM应用的新表示和指纹也正在开发中[127,128]。鉴于基于DNN的模型在其他计算化学领域的传统机器学习模型中观察到的高精度,我们建议开发基于DNN的模型 机器学习QM应用的例子对该领域有利。
计算材料设计
量子化学领域的DNN应用的逻辑扩展是预测和设计与QM计算的特性相关或基于QM计算的特性的材料特性。定量结构特性关系(QSPR),是QSAR在非生物领域的类似版本,是预测物理性质的科学,其从更基本的物理化学特性出发,在之前的出版物中得到了广泛的综述。[129,130 ]与现代药物开发早期相似,物质发现主要是由偶然性和机构记忆驱动的[131]。这使得该领域成为探索性的试验方法,而分子材料设计的关键瓶颈在于实验的合成和表征。近年来,计算和合理材料设计的范例已被封装在材料基因组计划下[132,133]。由于这一领域的新颖性,在本节中,我们将研究使用计算材料的机器学习的一些关键成就设计并突出深度学习应用场景。
Raccuglia等人发表了一篇最近使用机器学习模型加速材料性能研究的高调例子。在2016年。[30]无机 - 有机杂化材料如金属有机骨架(MOFs)的合成已经被广泛研究了数十年,但对这些化合物形成的理论理解仅部分被理解。在Raccuglia等人的工作中,作者使用基于SVM的模型来预测模板化钒亚硒酸盐结晶的反应结果。关于他们的工作有趣的是,在训练模型时纳入了“黑暗”反应,这些反应是从存档的实验室笔记本收集的失败或不成功的反应。由目标化合物类型的合成所定义的,所得模型具有89%的成功率。值得注意的是,这超过了78%的人类直觉成功率[131]。虽然在本研究中没有使用基于DNN的模型,但没有技术上的原因说明它不能用来代替SVM作为用于计算材料合成预测的工具。
2015年,Aspuru-Gizik及其同事报告了DNN如何用于加速材料发现的一个例子。[134]在这里,作者使用从哈佛清洁能源项目获得的数据集 - 高通量虚拟筛选工作,发现高性能有机光伏材料。要预测的度量是功率转换效率(PCE),其是HOMO和LUMO能量以及几个其他经验参数的函数。[134]由于没有高质量的三维数据可用于生成库仑矩阵,作者决定使用基于分子图的指纹作为输入表示。测试了四种不同的表示,结果显示HOMO,LUMO和PCE预测的准确性一致(在同一数量级内)。该数据集由从CEPDB数据库中随机选择的200,000种化合物组成,另有50,000种被提取作为测试集。 HOMO和LUMO的测试误差分别为0.15和0.12eV,这与von Lilienfeld及其同事开发的DNN模型相当。
虽然材料设计中的DNN应用仍处于起步阶段,但看看它的应用如何应对传统QSPR应用和即将进行的合理材料设计工作(如预测荧光团的光谱特性[135,136]、离子液体的特性[137]、和纳米结构的活性[138]))将会很有趣。
关于深度学习和黑盒子特性的保守看法
机器学习算法虽然可能不是我们领域许多从业人员首选的工具,但无可否认在化学信息学领域以及QSAR和蛋白质结构预测等应用领域有着丰富的历史。虽然有人可能认为深度学习在某种意义上是以前的人工神经网络的复兴,但过去十年的算法和技术突破使得开发出惊人复杂的深度神经网络,允许训练具有数亿权重。加上数据和GPU加速科学计算的发展,深度学习推翻了计算机科学领域的许多应用,如语音识别和计算机视觉。鉴于化学领域中类似的相似之处,这表明深度学习可能是一个有价值的工具,可以添加到计算化学工具箱中。正如表1所总结的那样,它提出了基于DNN模型的关键初步出版物,我们注意到深度学习在计算化学许多子领域的广泛应用。此外,基于DNN的模型的性能几乎总是等同于现有的最先进的非神经网络模型,并且有时提供了优异的性能。然而,我们注意到,如果要比较DNN为其语音识别和计算机视觉的“母体”领域带来的改进,许多情况下的性能提升并不显着。解释化学领域缺乏革命性进展的一个缓解因素可能是数据的相对稀缺性。与数据便宜的计算机科学领域不同,尤其是从互联网或社交媒体获得的数据时,由于需要进行实际的实验或计算以生成有用的数据,所以化学中可用数据的数量可以理解为更小且更昂贵。此外,化学领域已经存在了几个世纪,并且考虑到化学原理基于物理定律的事实,例如分子描述符等特征的发展来解释化合物溶解度是不难想象的,例如,比开发功能来解释狗和猫之间的差异更容易,这是计算机视觉中的一项常见任务。因此,在化学中具有更精确和更好的工程特征,我们也可能看不到如此大的初始性能改进,尤其是对于相对简单的化学原理或概念。
此外,作为计算化学家,与工程师或技术人员相比,更重视概念理解,这可以说是计算机科学领域中比较流行的思维模式。 在这方面,深度学习算法目前在两个账户上不足。 首先,它缺乏基于实际物理定律的第一原理模型的概念优雅,第二,DNN本质上是一个黑盒子; 很难理解神经网络“已经学会”什么,或者它究竟如何预测感兴趣的属性。
为了解决概念优雅的第一个问题,从某种角度来看,这种反对可能更多地是科学偏好的哲学论证。在大多数计算化学应用中,除非人们精确地求解薛定谔方程,我们知道除了双体系统外其他方法都不可能,我们必须对模型进行近似。从这个意义上说,几乎所有的计算化学都是凭经验确定的,有时甚至直观地确定了薛定谔方程的“真实”第一性原理的近似值。为了说明这一点,让我们来看看古典分子模型力场的历史发展,如CHARMM [42]和AMBER。[43]例如,二面角力常数的参数化在历史上一直针对QM计算值,即以经验证的物理原理为基础的“真实”值。然而,由于真实分子的动力学行为不具有叠加作用(这本身就是经典分子模型的另一种近似),最近的重新参数化已经开始修改二面角参数,以经验拟合实验NMR分布,尽管这可能导致偏差[142,143]同样,模拟静电力的伦琴相互作用的选择也只是近似正确的,模型带电离子相互作用的最近参数开始已经开始拟合各种实验观察值,例如渗透压值,以及在建模特定的静电相互作用对时引入非物理修正项[144-146]在这些例子中,必须从第一原理进行逼近,而这个过程是一个基于经验数据或有时“化学直觉“ - 就像Raccuglia等人。已经表明,不是绝对可靠的,并不总是更准确。[131]在计算化学家所做的工作过于简单化的风险下,现有计算化学模型的发展可能被视为一种精细的曲线拟合练习。与其使用人类专家知识,可能的替代方案可能是使用深度学习算法来“建议”,或者甚至可能帮助我们“决定”应该做出什么样的近似以达到期望的结果,以朝着未来的范式转变基于DNN的人工智能(AI)辅助化学研究。这自然会导致深层学习的第二个缺点 - 不可避免的问题 - 我们如何知道深度学习模型正在学习正确的物理或化学?
我们会承认,在目前的实施中,深度学习算法仍然是一个黑匣子,并且询问它“学习”的内容是一项极具挑战性的任务。尽管如此,诸如SVM和RF之类的黑盒算法也被用于几种计算化学应用中,特别是在主要用作工具的示例中,和/或用于预测如此复杂的属性,以至于即使对于问题不一定有助于其预测。我们承认,要推动深入学习不仅仅是化学家工具包中的另一个工具,并且为了获得更广泛的适用性和科学研究的采用,显然DNN的可解释性的提高是最重要的。虽然神经网络的可解释性历来不是这一领域的从业人员强烈的研究焦点,但值得注意的是,近期有关提高可解释性的一些发展已有报道[147,148]。其他可行的选择包括使用不同的基于神经网络的机器学习模型,如为解释性而设计的影响相关性选民(IVR)。正如Baldi及其同事所做的一些计算化学应用所证明的,IRV是一种低参数神经网络,通过非线性地结合化学邻居在训练集中的影响来改进k-最近邻分类器。 IRV影响也被非线性地分解为相关成分和投票成分。因此,IRV的预测本质上是透明的,因为通过检查每个预测的影响可以从网络中提取用于进行预测的确切数据,使其更接近“白盒”神经网络方法[149,150]
结论
与目前计算化学中使用的传统机器学习算法不同,深度学习在其使用非线性函数的层次级联中有所区别。这使得它可以学习表示并从预测理想的物理化学性质所需的原始未处理数据中提取出必要的特征。正是这一特点使得深度学习在其语音识别和计算机视觉的“母体”领域中产生了重大影响和变革性影响。在计算化学中,其影响更近,更具初步性。尽管如此,根据近期一些研究的结果,我们注意到深度学习在许多计算化学领域的广泛应用,包括计算机辅助药物设计,计算结构生物学,量子化学和材料设计。在我们所研究的几乎所有应用中,基于DNN的模型的性能常常优于传统的机器学习算法。
随着问题复杂性的增加,能够应用多任务学习(即需要更多的不同属性的预测),随着数据集大小的增加,我们也看到了深入的学习从经常表现出色到始终优于传统机器学习模型。此外,一些初步研究结果表明,诸如分子描述符等明确设计的特征可能不需要构建高性能DNN模型,并且以分子指纹或库仑矩阵形式的简单表示可能就足够了。这是因为DNN能够通过隐藏层提取出自己的特征。甚至有迹象表明DNN“学习”的特征符合实际的化学概念,如毒素。加上最近关于提高神经网络可解释性的研究,它表明DNN在计算化学中的未来作用可能不仅仅是一种高性能的预测工具,而且可能也是一种假设生成装置。
参考文献
Goh G B, Hodas N O, Vishnu A. Deep learning for computational chemistry[J]. Journal of Computational Chemistry, 2017, 38(16):1291-1307.
文章来源: drugai.blog.csdn.net,作者:DrugAI,版权归原作者所有,如需转载,请联系作者。
原文链接:drugai.blog.csdn.net/article/details/105683669

- 点赞
- 收藏
- 关注作者
评论(0)