Python生物信息学③提取差异基因

【摘要】 python做生信分析的流程
使用的数据集是GSE5583,来自于2006年的基因芯片结果,该芯片目的是提取野生型和HDAC1小鼠胚胎干细胞用于Affymetrix微阵列上的差异RNA。
#导入包import matplotlib.pyplot as pltimport osimport numpy as npimport pandas as pdfrom scipy i...
python做生信分析的流程
使用的数据集是GSE5583,来自于2006年的基因芯片结果,该芯片目的是提取野生型和HDAC1小鼠胚胎干细胞用于Affymetrix微阵列上的差异RNA。
-
#导入包
-
import matplotlib.pyplot as plt
-
import os
-
import numpy as np
-
import pandas as pd
-
from scipy import stats
-
import seaborn as sns
-
%matplotlib inline
-
#载入数据
-
data = pd.read_table("GSE5583.txt",header = 0,index_col = 0)
-
data.head() #查看前5行
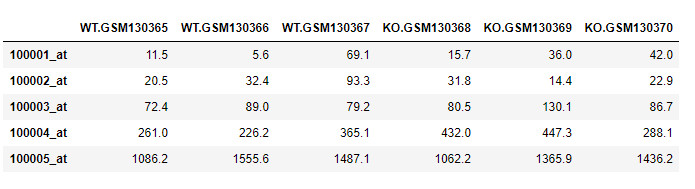
每一行是一个基因,每一列是一个样本,这也是比较经典的芯片数据集
-
#查看数据维度
-
data.shape
标准化
常见的log2()标准化
-
data2 = np.log2(data+0.0001)
-
data2.head()
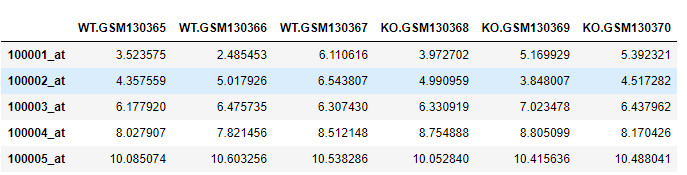
-
# 每个阵列的箱线图
-
plt.show(data2.plot(kind = 'box', title = 'GSE5583 Boxplot', rot = 90))
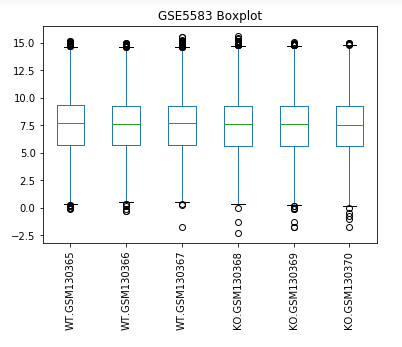
目的是查看不同样本之间是否有总体差异。
-
# Density
-
plt.show(data2.plot(kind = 'density', title = 'GSE5583 Density'))
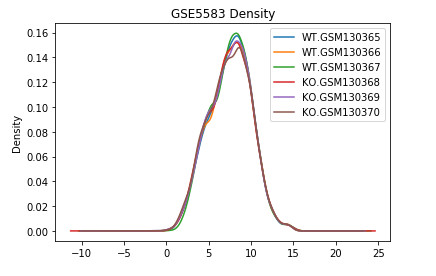
可以看出样本之间没有总体差异,可以做差异分析。
-
#每个基因(行)wt样本的表达平均值
-
wt = data2.loc[:, 'WT.GSM130365' : 'WT.GSM130367'].mean(axis = 1)
-
wt.head()
-
#每个基因(行)的ko样本的表达平均值
-
ko = data2.loc[:,'KO.GSM130368':'KO.GSM130370'].mean(axis = 1)
-
ko.head()
-
fold = ko - wt
-
-
#折叠变化的直方图
-
plt.hist(fold)
-
plt.title("Histogram of fold-change")
-
plt.show()
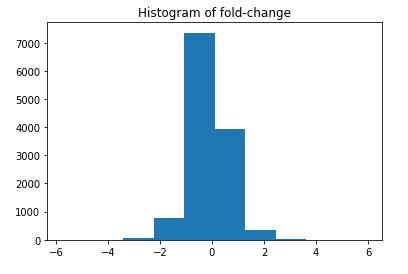
查看基因差异的P值分布
-
from scipy import stats
-
-
pvalue = []
-
for i in range(0, number_of_genes):
-
ttest = stats.ttest_ind(data2.iloc[i,0:3], data2.iloc[i,3:6])
-
pvalue.append(ttest[1])
-
-
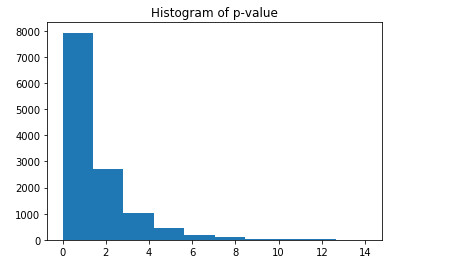
# Histogram of the p-values
-
plt.hist(-np.log(pvalue))
-
plt.title("Histogram of p-value")
-
plt.show()
参考:
https://www.jianshu.com/p/91c98585b79b
文章来源: drugai.blog.csdn.net,作者:DrugAI,版权归原作者所有,如需转载,请联系作者。
原文链接:drugai.blog.csdn.net/article/details/88877273

【版权声明】本文为华为云社区用户转载文章,如果您发现本社区中有涉嫌抄袭的内容,欢迎发送邮件进行举报,并提供相关证据,一经查实,本社区将立刻删除涉嫌侵权内容,举报邮箱:
cloudbbs@huaweicloud.com
- 点赞
- 收藏
- 关注作者
评论(0)