[数据科学] 通过基因表达监测进行肿瘤预测

简介
通过基因表达监测(DNA微阵列)对新的癌症病例进行分类,从而为鉴定新的癌症类别和将肿瘤分配到已知类别提供了一般方法。这些数据用于对患有急性髓性白血病(AML)和急性淋巴细胞白血病(ALL)的患者进行分类。
代码实例
导入依赖库
-
import numpy as np
-
import pandas as pd
-
import matplotlib.pyplot as plt
-
%matplotlib inline
载入数据
-
labels_df = pd.read_csv('actual.csv', index_col = 'patient')
-
test_df = pd.read_csv('data_set_ALL_AML_independent.csv')
-
data_df = pd.read_csv('data_set_ALL_AML_train.csv')
-
print('train_data: ', data_df.shape, '\n test_data: ', test_df.shape, '\n labels: ', labels_df.shape)
-
# labels_df.shape
-
data_df.head() #查看前5行(默认前5行)
清理数据
-
test_cols_to_drop = [c for c in test_df.columns if 'call' in c]
-
test_df = test_df.drop(test_cols_to_drop, axis=1)
-
test_df = test_df.drop(['Gene Description', 'Gene Accession Number'], axis=1 )
-
-
data_cols_to_drop = [c for c in data_df.columns if 'call' in c]
-
data_df = data_df.drop(data_cols_to_drop, axis=1)
-
data_df = data_df.drop(['Gene Description', 'Gene Accession Number'], axis=1 )
-
print('train_data ', data_df.shape, '\n test_data: ', test_df.shape, '\n labels: ', labels_df.shape)
-
data_df.head()
定义'特征'和'样本'
使用基因表达值来预测癌症类型。 因此,特征是患者的基因和样本。 使用X作为输入数据,其中行是样本(患者),列是特征(基因)。
将'ALLL'替换为0,将'AML'替换为1。
-
labels_df = labels_df.replace({'ALL':0, 'AML':1})
-
train_labels = labels_df[labels_df.index <= 38]
-
test_labels = labels_df[labels_df.index > 38]
-
print(train_labels.shape, test_labels.shape)
-
# labels_df.index
-
test_df = test_df.T
-
train_df = data_df.T
检查空值
print('Columns containing null values in train and test data are ', data_df.isnull().values.sum(), test_df.isnull().values.sum())
联合训练集和测试集
-
full_df = train_df.append(test_df, ignore_index=True)
-
print(full_df.shape)
-
full_df.head()
标准化和处理高维度
标准化
所有变量具有非常相似的范围的方式重新调整我们的变量(预测变量)。
高维度
只有72个样本和7000多个变量。 意味着如果采取正确的方法,模型很可能会受到HD的影响。 一个非常常见的技巧是将数据投影到较低维度空间,然后将其用作新变量。 最常见的尺寸减小方法是PCA。
-
# Standardization
-
from sklearn import preprocessing
-
X_std = preprocessing.StandardScaler().fit_transform(full_df)
-
# Check how the standardized data look like
-
gene_index = 1
-
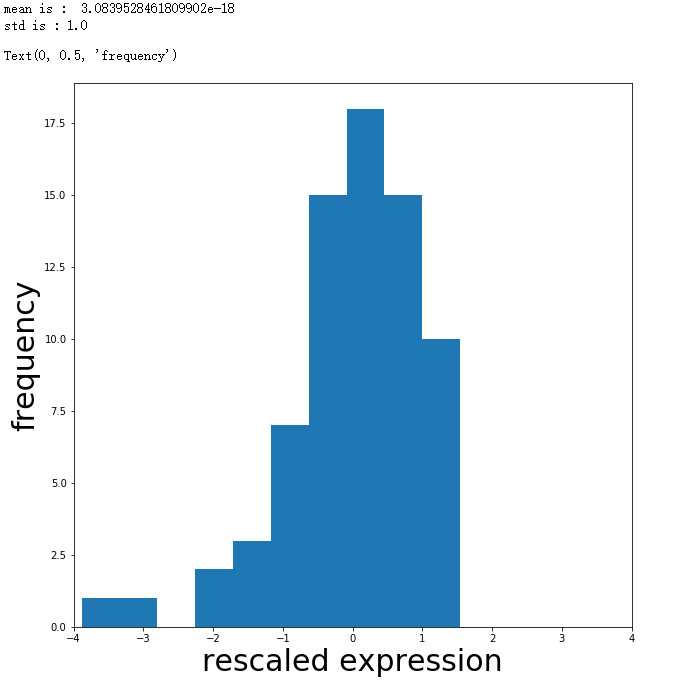
print('mean is : ', np.mean(X_std[:, gene_index] ) )
-
print('std is :', np.std(X_std[:, gene_index]))
-
-
fig= plt.figure(figsize=(10,10))
-
plt.hist(X_std[:, gene_index], bins=10)
-
plt.xlim((-4, 4))
-
plt.xlabel('rescaled expression', size=30)
-
plt.ylabel('frequency', size=30)
PCA(聚类分析)
-
# PCA
-
-
from sklearn.decomposition import PCA
-
pca = PCA(n_components=50, random_state=42)
-
X_pca = pca.fit_transform(X_std)
-
print(X_pca.shape)
-
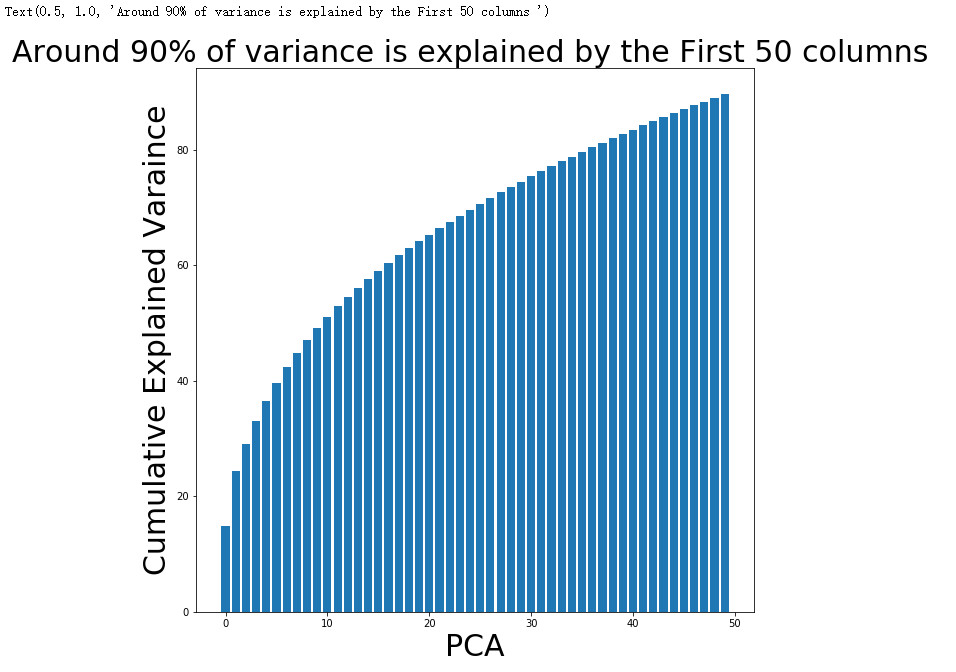
cum_sum = pca.explained_variance_ratio_.cumsum()
-
cum_sum = cum_sum*100
-
-
fig = plt.figure(figsize=(10,10))
-
plt.bar(range(50), cum_sum)
-
plt.xlabel('PCA', size=30)
-
plt.ylabel('Cumulative Explained Varaince', size=30)
-
plt.title("Around 90% of variance is explained by the First 50 columns ", size=30)
-
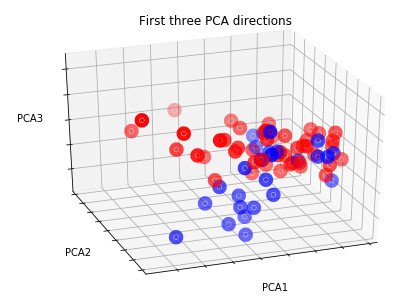
labels = labels_df['cancer'].values
-
-
colors = np.where(labels==0, 'red', 'blue')
-
-
-
from mpl_toolkits.mplot3d import Axes3D
-
plt.clf()
-
fig = plt.figure(1, figsize=(15,15 ))
-
ax = Axes3D(fig, elev=-150, azim=110,)
-
ax.scatter(X_pca[:, 0], X_pca[:, 1], X_pca[:, 2], c=colors, cmap=plt.cm.Paired,linewidths=10)
-
ax.set_title("First three PCA directions")
-
ax.set_xlabel("PCA1")
-
ax.w_xaxis.set_ticklabels([])
-
ax.set_ylabel("PCA2")
-
ax.w_yaxis.set_ticklabels([])
-
ax.set_zlabel("PCA3")
-
ax.w_zaxis.set_ticklabels([])
-
plt.show()
RF分类
-
X = X_pca
-
y = labels
-
print(X.shape, y.shape)
划分训练集和测试集
-
from sklearn.model_selection import train_test_split
-
-
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.33, random_state=42)
-
print(X_train.shape, y_train.shape)
-
import seaborn as sns
-
from sklearn.ensemble import RandomForestClassifier
-
from sklearn.metrics import accuracy_score
-
rfc = RandomForestClassifier(random_state=42)
-
rfc.fit(X_train, y_train)
-
y_pred = rfc.predict(X_test)
-
print(accuracy_score(y_test, y_pred))
0.7083333333333334
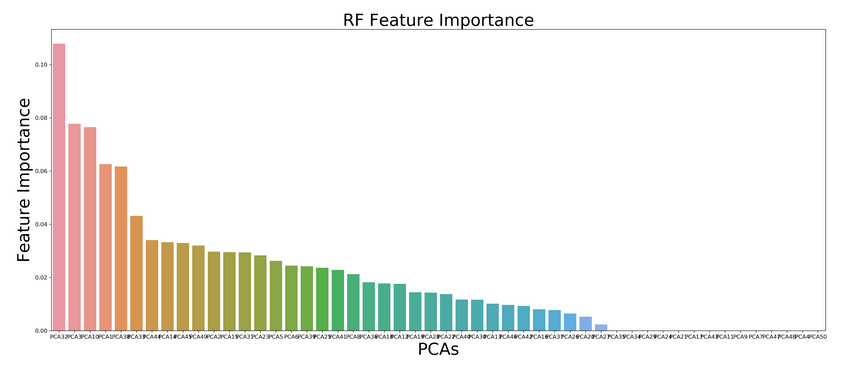
重要特征
-
labs = ['PCA'+str(i+1) for i in range(X_train.shape[1])]
-
importance_df = pd.DataFrame({
-
'feature':labs,
-
'importance': rfc.feature_importances_
-
})
-
-
importance_df_sorted = importance_df.sort_values('importance', ascending=False)
-
importance_df_sorted.head()
-
-
fig = plt.figure(figsize=(25,10))
-
sns.barplot(data=importance_df_sorted, x='feature', y='importance')
-
plt.xlabel('PCAs', size=30)
-
plt.ylabel('Feature Importance', size=30)
-
plt.title('RF Feature Importance', size=30)
-
plt.savefig('RF Feature Importance.png', dpi=300)
-
plt.show
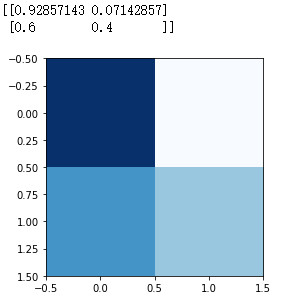
Confusion Matrix(混淆矩阵)
Gradient Boost Classifier(梯度增强分类器)
-
from sklearn.metrics import confusion_matrix
-
cm = confusion_matrix(y_test, y_pred)
-
### Normalize cm, np.newaxis makes to devide each row by the sum
-
cm = cm.astype('float') / cm.sum(axis=1)[:, np.newaxis]
-
-
-
print(np.newaxis)
-
cmap=plt.cm.Blues
-
-
plt.imshow(cm, interpolation='nearest', cmap=cmap)
-
print(cm)
-
from sklearn.ensemble import GradientBoostingClassifier
-
gbc = GradientBoostingClassifier(random_state=42)
-
gbc.fit(X_train, y_train)
-
y_gbc_pred = gbc.predict(X_test)
-
print(accuracy_score(y_test, y_gbc_pred))
0.7083333333333334
参考:
Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Expression
Science 286:531-537. (1999). Published: 1999.10.14
T.R. Golub, D.K. Slonim, P. Tamayo, C. Huard, M. Gaasenbeek, J.P. Mesirov, H. Coller, M. Loh, J.R. Downing, M.A. Caligiuri, C.D. Bloomfield, and E.S. Lander
文章来源: drugai.blog.csdn.net,作者:DrugAI,版权归原作者所有,如需转载,请联系作者。
原文链接:drugai.blog.csdn.net/article/details/99676017

- 点赞
- 收藏
- 关注作者
评论(0)