Indigo | Indigo(Python)简介、安装与入门

【摘要】 Indigo简介
Bingo: 针对Oracle,Microsoft SQL Server和PostgreSQL数据库的化学搜索引擎Indigo: U具有与.NET,Java和Python绑定的通用化学信息库,以及以下工具:Legio: 组合化学GUI应用程序ChemDiff: SDF或SMILES文件的可视化比较indigo-depict: 分子和反应渲染工具indigo...
Indigo简介
- Bingo: 针对Oracle,Microsoft SQL Server和PostgreSQL数据库的化学搜索引擎
- Indigo: U具有与.NET,Java和Python绑定的通用化学信息库,以及以下工具:
- Legio: 组合化学GUI应用程序
- ChemDiff: SDF或SMILES文件的可视化比较
- indigo-depict: 分子和反应渲染工具
- indigo-cano: Canonical SMILES 生成器
- indigo-deco: R-Group反卷积实用程序
Indigo安装
Indigo的Python绑定安装
pip install epam.indigo
Indigo入门
-
from indigo import *
-
-
indigo = Indigo()
获取版本
print ("Indigo version " + indigo.version())
Accessing Neighbor Atoms
-
for atom in mol.iterateAtoms():
-
print ("atom %d: %d neighbors" % (atom.index(), atom.degree()))
-
for nei in atom.iterateNeighbors():
-
print ("neighbor atom %d is connected by bond %d\n" % (nei.index(), nei.bond().index()))
Accessing R-Groups
-
for rg in mol.iterateRGroups():
-
print ("RGROUP #" + rg.index())
-
for frag in rg.iterateRGroupFragments():
-
print (" FRAGMENT #" + rg.index())
-
print (frag.molfile())
Stereochemistry
The following methods of IndigoObject are available for accessing molecule’s stereo configuration:
countStereocentersreturns the number of the chiral atoms in a moleculeiterateStereocentersreturns an iterator for molecule’s atoms that are stereocenterscountAlleneCentersreturns the number of allene-like stereo fragmentsiterateAlleneCentersreturns an iterator for molecule’s atoms that are centers of allene fragments (the middle ‘C’ in ‘C=C=C’)bondStereoreturns one of the following constants:Indigo.UP— stereo “up” bondIndigo.DOWN— stereo “down” bondIndigo.EITHER— stereo “either” bondIndigo.CIS— “Cis” double bondIndigo.TRANS— “Trans” double bond- zero — not a stereo bond of any kind
stereocenterTypereturns one of the following constants:Indigo.ABS— “absolute” stereocenterIndigo.OR— “or” stereocenterIndigo.AND— “and” stereocenterIndigo.EITHER— “any” stereocenter- zero — not a stereocenter
invertStereoinverts the stereo configuration of an atomresetStereoresets the stereo configuration of an atom or a bondchangeStereocenterType(newType)changes current stereocenter type to a specified typeaddStereocenter(type, idx1, idx2, idx3, [idx4])adds new stereocenter build on a atom pyramid with a specified atom indicesclearStereocentersresets the chiral configurations of a molecule’s atomsclearAlleneCentersresets the chiral configurations of a molecule’s allene-like fragmentsclearCisTransresets the cis-trans configurations of a molecule’s bonds
The following methods are useful for keeping cis-trans stereochemistry intact when converting to/from SMILES:
resetSymmetricCisTranscan be called on a molecule loaded from a Molfile or CML. After this call, the cis-trans configurations remain only on nonsymmetric cis-trans bonds. The method returns the number of bonds that have been reset.markEitherCisTranscan be called prior to saving a molecule loaded from SMILES to Molfile format. It guarantees that the bonds that have no cis-trans configuration in SMILES will not have a cis-trans configuration in the resulting Molfile.
-
IndigoObject mol = indigo.loadMolecule("chiral.mol");
-
-
print mol.countStereocenters(), "chiral atoms"
-
for atom in mol.iterateStereocenters():
-
print "atom", atom.index(), "-- stereocenter type", atom.stereocenterType()
-
atom.invertStereo();
-
-
for bond in mol.iterateBonds():
-
if bond.bondStereo() != 0:
-
print "bond", bond.index(), "-- stereo type", bond.bondStereo()
-
-
print mol.smiles()
-
mol.clearStereocenters()
-
mol.clearCisTrans()
-
print mol.smiles()
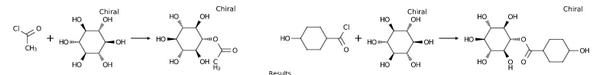
Reaction Products Enumeration
-
reaction = indigo.loadQueryReaction("Cl[C:1]([*:3])=O.[OH:2][*:4]>>[*:4][O:2][C:1]([*:3])=O")
-
-
monomers_table = indigo.createArray()
-
-
monomers_table.arrayAdd(indigo.createArray())
-
monomers_table.at(0).arrayAdd(indigo.loadMolecule("CC(Cl)=O"))
-
monomers_table.at(0).arrayAdd(indigo.loadMolecule("OC1CCC(CC1)C(Cl)=O"))
-
-
monomers_table.arrayAdd(indigo.createArray())
-
monomers_table.at(1).arrayAdd(indigo.loadMolecule("O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O"))
-
-
output_reactions = indigo.reactionProductEnumerate(reaction, monomers_table)
-
-
indigo.setOption("render-comment", "Results")
-
rxn_array = indigo.createArray();
-
for elem in output_reactions.iterateArray():
-
rxn = elem.clone();
-
rxn_array.arrayAdd(rxn)
-
-
indigoRenderer.renderGridToFile(rxn_array, None, 2, 'result_rpe.png')
Reaction-based Molecule Transformations
-
reaction = indigo.loadQueryReaction("[*+:1][*-:2]>>[*:2]=[*:1]")
-
molecule = indigo.loadMolecule("[O-][C+]1CCCC1[N+]([O-])=O")
-
indigo.transform(reaction, molecule)
-
print(molecule.smiles())
运行结果
O=N(C1CCCC1=O)=O
参考
1. https://github.com/epam/Indigo
2. https://lifescience.opensource.epam.com/
3. https://pypi.org/project/epam.indigo/
4. https://lifescience.opensource.epam.com/indigo/api/index.html
文章来源: drugai.blog.csdn.net,作者:DrugAI,版权归原作者所有,如需转载,请联系作者。
原文链接:drugai.blog.csdn.net/article/details/103662058

【版权声明】本文为华为云社区用户转载文章,如果您发现本社区中有涉嫌抄袭的内容,欢迎发送邮件进行举报,并提供相关证据,一经查实,本社区将立刻删除涉嫌侵权内容,举报邮箱:
cloudbbs@huaweicloud.com
- 点赞
- 收藏
- 关注作者
评论(0)