J. Cheminform. | GraphSol:预测接触图助力蛋白质溶解度预测


今天给大家介绍中山大学杨跃东教授课题组发表在Journal of Cheminformatics上的一篇论文。该论文指出蛋白质的溶解度对于生产新的可溶性蛋白质非常重要,但是目前预测蛋白质溶解度的方法大多基于氨基酸的一维嵌入,仅限于捕获空间结构信息。针对上述问题,作者开发了一种新的结构感知方法——GraphSol,通过注意力图卷积网络(GCN)预测蛋白质的溶解度,其中蛋白质拓扑结构属性图是通过预测序列的接触图构建的,结果显示GraphSol明显优于其他基于序列的方法。
1
介绍
近20年来,蛋白质的溶解度在生物技术和制药工业的蛋白质生产中起着重要作用。现有的预测蛋白质溶解度的计算方法主要是:基于物理的方法和基于机器/深度学习的方法。在基于物理的技术方面,大多数工作集中在利用分子动力学模拟来评估(聚集和溶解状态间)的自由能差,但这些方法通常准确性有限。而对于机器/深度学习技术,大部分方法主要基于LSTM和CNN,没有利用蛋白质分子的空间信息。
随着蛋白质结构预测的最新发展,蛋白质接触图的预测效果得到改善,预测的蛋白质接触图可以高精度地表示2D结构特征。在这项研究中,作者通过结合预测的接触图和图神经网络,以新颖的结构感知方法GraphSol预测蛋白质的溶解度。作者表明这是第一项通过图神经网络对蛋白质进行基于序列的溶解度预测的研究,并且该结构可以轻松应用于蛋白质的广泛任务中,例如蛋白质功能预测,蛋白质-蛋白质相互作用预测,蛋白质折叠和药物设计。
2
模型
这项工作中,作者将蛋白质溶解度预测任务转换为基于图的回归问题。给定一个由????个氨基酸组成的蛋白质序列,整个蛋白质因此可以表示为拓扑属性图????(????,????),其中F代表所有残基(节点)的特征集,????代表残基接触(边缘)。根据预测的蛋白质接触图,通过图卷积神经网络模型输出预测的溶解度。
GraphSol模型包括三个部分。如图1所示,第一部分是图卷积网络(GCN),它在迭代过程中从其节点和边缘聚合蛋白质结构信息。第二部分是一个自注意层,它将不同大小的节点隐藏状态转换为具有固定大小的图形表示向量。最后,这个固定大小向量将经过全连接层以预测蛋白质的溶解度。
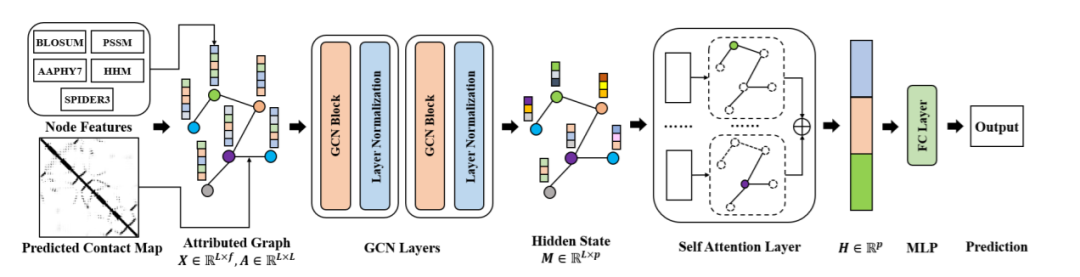
图1 GraphSol
3
实验
数据集
为了训练模型,作者使用了eSOL数据集,共有2737种蛋白质序列。从这些序列中,随机选择75%(2052个蛋白)作为训练集,其余685个蛋白用作独立测试。对于外部独立测试,作者选择了从酿酒酵母中收集的另一个蛋白质数据集。该数据集是通过包含108种具有相应3D结构的蛋白质而得出的。数据集考虑了如表1所示的四种节点特征。
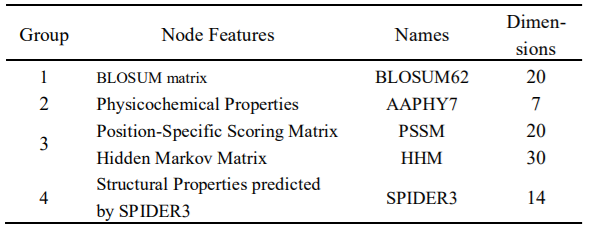
表1 数据集节点特征
实验结果
作者测试了eSOL数据集上GraphSol模型的性能。如表2所示,对于5折CV和独立测试,获得的R2值分别为0.476±0.014和0.483。当通过0.5的临界值将数据集分为两个离散状态(可溶或不可溶)时,对于5折CV和独立测试,AUC值分别为0.855和0.866, CV和独立测试的相似结果表明了GraphSol模型的鲁棒性。为了表明特征组的重要性,作者在消融研究中通过3种方式评估了性能。如表2所示,当将单个要素组用作节点要素时,在独立测试中,HHM产生的R2最高。预测的结构特征组(SPIDER3)的性能最差。相反,当去除单个基团时,去除SPIDER3导致最大的下降,这可能是因为SPIDER3提供了唯一的结构信息,而其他功能则具有补充功能。当通过递归添加特征组来评估模型时,模型会随着每个特征组的添加而显示出递增的性能。添加SPIDER3特征后,性能迅速提高,这反映了溶解度和结构特征之间的关系。
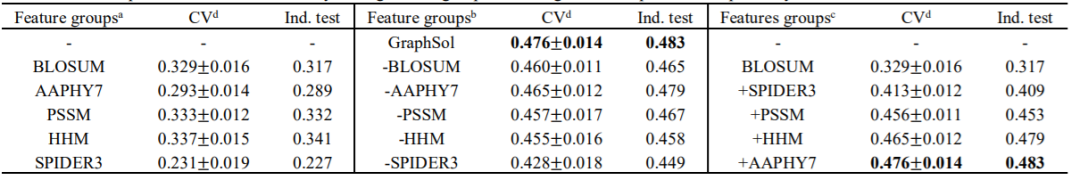
表2 实验结果
作者将GraphSol模型与最新方法进行了比较。为了避免不同数据集的影响,分别对所有机器学习或基于深度学习的模型进行了重新训练,并分别在训练集和测试集上进行了测试。如表3所示,GraphSol始终获得最佳结果。
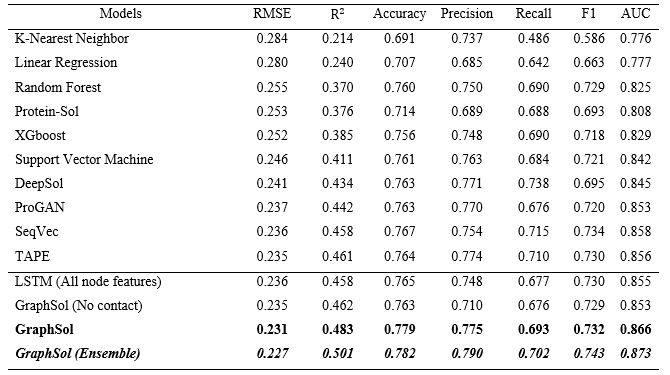
表3 方法比较
4
总结
这项研究中,作者提出了一种基于序列的预测蛋白质溶解度的方法GraphSol。与其他方法相比,GraphSol利用了预测的蛋白质接触图,该图在连接蛋白质拓扑属性和注意力图神经网络中发挥了关键作用。作者指出,残基之间的预测接触概率比离散状态更能表示成对关系,这种方法在蛋白质功能预测、蛋白质相互作用预测、蛋白质折叠和药物设计等蛋白质属性预测方面具有潜在的应用价值。
参考资料
Chen, J., Zheng, S., Zhao, H. et al. Structure-aware protein solubility prediction from sequence through graph convolutional network and predicted contact map. J Cheminformatics 13, 7 (2021).
https://doi.org/10.1186/s13321-021-00488-1
文章来源: drugai.blog.csdn.net,作者:DrugAI,版权归原作者所有,如需转载,请联系作者。
原文链接:drugai.blog.csdn.net/article/details/114650426

- 点赞
- 收藏
- 关注作者
评论(0)